





Introducción. La hemocromatosis hereditaria (HH) es una enfermedad autosómica recesiva. La principal mutación es C282Y, aunque hay otras como la H63D1. La prevalencia es aproximadamente un 10% de la población para heterocigotos y un 0,5% para homocigotos en las poblaciones más afectadas, como la caucásica estadounidense y del oeste de Europa. Es una afección multiorgánica en que se acumula hierro en las células parenquimatosas de hígado, piel, corazón y páncreas.
Caso clínico. Paciente de 38 años que acude al centro de salud por dolor en el carpo de meses de evolución, sin eritema, calor ni traumatismo previo. Antecedentes personales de meningitis a los 9 años, hiperuricemia desde hace aproximadamente 20 años; no bebedor, fumador de 15-20 cigarrillos/día. Yesista. Dentro de sus antecentes familiares destaca su padre con enfermedad por depósito de hierro, sin estudiar.
Se realiza analítica con Hb, 14,5; HCT, 44,3; VCM, 88,6; HCM, 29,3; CHCM, 33; plaquetas, 230.000; glucosa basal, 148; creatinina, 1,2; ácido úrico, 8,5; colesterol total, 213; GOT, 39; GPT, 50; G-GT, 142; albúmina, 4,6; bilirrubina total, 0,6; fosfatasa alcalina, 136; orina, normal. Observando alteraciones analíticas de glucosa basal, 121 mg/dl; hierro sérico, 178; saturación transferrina (IST), 54% y ferritina, 383. Posteriormente, el IST llegó al 66%, realizándose biopsia hepática (especialista): hemosiderosis intracitoplásmica escasa. En el estudio genético realizado en el centro de salud se detecta la mutación H63D heterocigoto.
Se comienza con flebotomías periódicas (hasta 3 en 6 meses), consiguiendo glucosa, 105; úrico, 9,1; GOT, 36; CPT, 41; GGT, 114, ferritina, 8,07 ng/ml; hierro, 26; con el paciente totalmente asintomático.
Discusión y conclusiones. En los últimos años se ha ido descubriendo el genotipo de las HH a través de estudios2,3, entre el 91 y el 64% son homocigotos C282Y, los claramente afectados, pero hay un porcentaje de entre el 3 y el 4% con genotipo H63D/alelo normal, con menor depósito de hierro en tejidos y algunos de ellos, esteatohepatitis no alcohólica, bebedores, porfiria cutánea tarda y otros ninguna otra alteración, como nuestro caso. Es curioso que el alelo mutante por vía paterna, puede producir una mayor expresión de la enfermedad que por vía materna4.
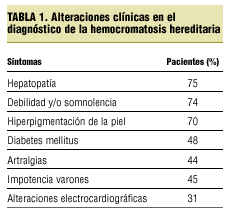
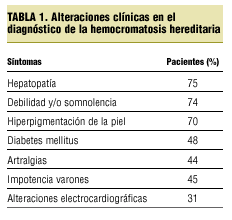
La forma de presentación varía (tabla 1), aunque sí es cierto que una hiperuricemia justifica una artritis de carpo, la clínica no era una clara artritis microcristalina; además, con las flebotomías desapareció.
Con las flebotomías, nuestro paciente mejoró en meses, normalizándose todos sus valores y manteniendo ferritina menor de 10 ng/ml sin provocar anemia con flebotomías cada 4-6 meses, debido a la menor sobrecarga de hierro que en pacientes C282Y/C282Y5, igualándose la expectativa de vida en los pacientes que están en fases tempranas de la enfermedad con sobrecarga leve de hierro con la de pacientes sanos6.
