


Diabetes is one of the few growing causes of a cardiomyopathy around the world and cardiomyopathy is a common complication of diabetes causing significant mortality and morbidity.
This cardiomyopathy is seen commonly in young people with diabetes and those without pre-existing coronary artery disease, suggestive of a specific entity linked directly to diabetes.
In recent years there has been a real upsurge in the research devoted to this growing problem as other causes of heart disease such as tobacco smoking wane. More and more animal model studies and human tissue studies have enabled researchers to begin to develop ideas of the processes causing this cardiomyopathy.
As the pathological processes causing this cardiomyopathy are beginning to be better understood, we present an overview of the various potential pathological mechanisms under investigation that may constitute a cardiomyopathy related to diabetes.
In our review we describe 6 possible processes, which may begin to explain the cardiomyopathy related to diabetes beyond the standard ischaemia-infarct model. These mechanisms which are still under investigation include, reduced metabolic function, reactive oxygen species damage, damage to the ryanodine receptor, up-regulation of ADH, a cardiac autonomic neuropathy, and changes in cardiac structure.
La diabetes es una de las escasas causas crecientes de miocardiopatía en el mundo, y la miocardiopatía es una complicación común de la diabetes que provoca una importante morbilidad y mortalidad.
Tal miocardiopatía se observa habitualmente en personas jóvenes con diabetes y sin coronariopatía previa, lo que sugiere una entidad específica relacionada directamente con la diabetes.
Los últimos años han sido testigos de una oleada de investigaciones dedicadas a este creciente problema, a medida que disminuían otras causas de cardiopatía, como el tabaquismo. Una gran cantidad de trabajos sobre modelos animales y sobre tejido humano han permitido a los investigadores iniciar el desarrollo de una idea sobre los procesos que causan esta miocardiopatía.
Dado que se comienza a conocer mejor los procesos anatomopatológicos que provocan esta miocardiopatía, presentamos una revisión de los distintos posibles mecanismos anatomopatológicos en investigación que podrían desembocar en una miocardiopatía relacionada con la diabetes.
Nuestra revisión describe 6 posibles procesos que podrían empezar a explicar la miocardiopatía relacionada con la diabetes más allá del modelo habitual de isquemia-infarto. Estos mecanismos, todavía en investigación, son la disminución de la función metabólica, la lesión por radicales oxígeno, el daño al receptor de rianodina, la regulación al alza de la ADH, una neuropatía del sistema autónomo cardíaco y alteraciones en la estructura cardíaca.
The association between diabetes and coronary heart disease is widely recognised, but diabetes as a cause of heart failure, irrespective and independent of coronary disease, has been the subject of much research. In 1972, Rubler and co-workers first described diabetic cardiomyopathy in 4 patients with diabetes presenting with clinical heart failure in the absence of hypertension, coronary or structural heart disease.1
In the UK, the estimated prevalence of diabetes is 4.3% while the prevalence of diabetes in people with congestive heart failure is quoted as between 20 and 35%per cent reflecting the earlier work in Framingham.2
There are many other epidemiological associations between diabetes and heart failure. In the Framingham study, there was an increased incidence of congestive cardiac failure in people with diabetes, which was independent of age, hypertension, hyperlipidaemia and coronary heart disease.3 The presence of hypertension, coronary artery disease and systolic impairment conferred a much poorer prognosis in these patients. People with diabetes are also more likely than those without, to develop heart failure following myocardial infarction even when infarct size is comparable.4 Heart failure symptoms with a preserved ejection fraction, as can be seen in many patients with diabetes, carries a significant mortality risk.5
Healthy, asymptomatic patients with diabetes have been shown to demonstrate subtle differences in systolic and diastolic function. Children with type 1 diabetes have been found to have subtle changes suggestive of cardiac impairment, including impaired myocardial relaxation patterns, a phenomenon that was previously confirmed in adult diabetes studies.6
In this review we will look at what is the current thinking on the pathological mechanisms causing diabetic cardiomyopathy so we can understand the pathology of a process that affects so many patients with diabetes and which carries such a significant impact on their mortality and quality of life.
In this review we will focus on six potential pathological mechanisms that are currently the subject of much research both in human and animal studies. While there may be a difference between the two, animal work has often proved illustrative of the human processes.
Cardiac Energy Usage and ConsumptionThe heart is one of the most energy intense organs in the body and uses a between two- thirds to one-third ratio of free fatty acids to pyruvate for energy metabolism.
This careful balance is altered in people with diabetes and leads to a reduction in the heart's ability to meet its energy requirements and thus leads to heart failure.7 Glucose utilisation is vital in maintaining efficient energy production and also in protecting against periods of ischaemia8 and in diabetes glucose utilisation is impaired.
The key areas affected in diabetes seem to surround GLUT-4 and an excess of fatty acids for metabolism and we will review these areas now.
In all patients with heart failure, there is a reduction in the expression of the GLUT-4 receptor, the main glucose transporter in the heart, which causes reduced energy production. Down regulation of GLUT-4 expression in diabetes occurs through a reduction in the expression of myocyte enhancer factor 2C (MEF2C), which is a regulatory factor for GLUT-4 and key to ongoing production.9 In diabetes specifically, glucose metabolism seems to be also reduced due a reduction in the translocation of GLUT-4 to where it is needed, which seems to be the main difference between people with diabetes and heart failure and those without.8
As alluded to above, fatty acids in excess lead to reduced glucose utilisation and therefore reduced energy production, and in animal models excess free fatty acid metabolism leads to inhibition of phosphofructokinase, a rate-limiting step in glycolysis.10 Adipocytes that generate the offending excess free fatty acids become resistant to negative signalling in diabetes and help down regulate insulin mediated glucose uptake.11
Above we have looked at how glucose metabolism is inhibited and therefore the heart's energy demands cannot be met, but fatty acids do not just affect glucose usage, they are toxic in themselves. In patients with diabetes and diabetic animal models, an excess of fatty acids and triglycerides leads to the creation of toxic substrates which have a wide range of negative effects on cardiac function, including up-regulation of cell apoptosis.10 In animal models, the response to this excess of fats has been to increase CD36 and fatty acid transport protein (FATP) expression and their relocation to increase fatty acid uptake and oxidation.10 There is, therefore, not only production of toxic metabolites but diabetes leads to an increase in the absorption of the fatty acids that create a vicious cycle.
The changes we have seen so far that lead to impaired glucose utilisation and lipotoxic fatty acid production activate the peroxisome proliferator activated receptor-alpha system (PPAR Alpha). This has wide-ranging inhibitory effects on cardiac metabolism,12 via the peroxisome proliferator response element which increases the expression of the gene that increases fatty acid uptake and oxidation.13 Carnitine palmitoyltransferase I, an enzyme, which in normal people is strictly controlled but through PPAR-alpha activation in type 2 diabetes, is up-regulated and allows the excess fatty acid oxidation to continue unhindered.13
The long term effect of these changes is to impair contractile function, iso-volumetric relaxation and diastolic filling through insufficient energy supply and subsequently a reduction in the diabetic heart's ability to cope with ischaemia.10
Reactive Oxygen Species (ROS)One of the major factors in diabetic cardiomyopathy, as opposed to other forms of cardiomyopathy, is the constant production of damaging oxygen free radicals and their effects on the heart.
In times of free radical generation, the body acts through a variety of systems to respond to this, including free radical absorption (via NADPH and glutathione), repair to damaged structures and through planned apoptosis. This is a carefully balanced and well-adapted mechanism that helps the body in situations of infection, inflammation and damage from sunlight. It is becoming clearer that the diabetic state results in such a continuous stream of free radicals that the body's defences are unable to cope.
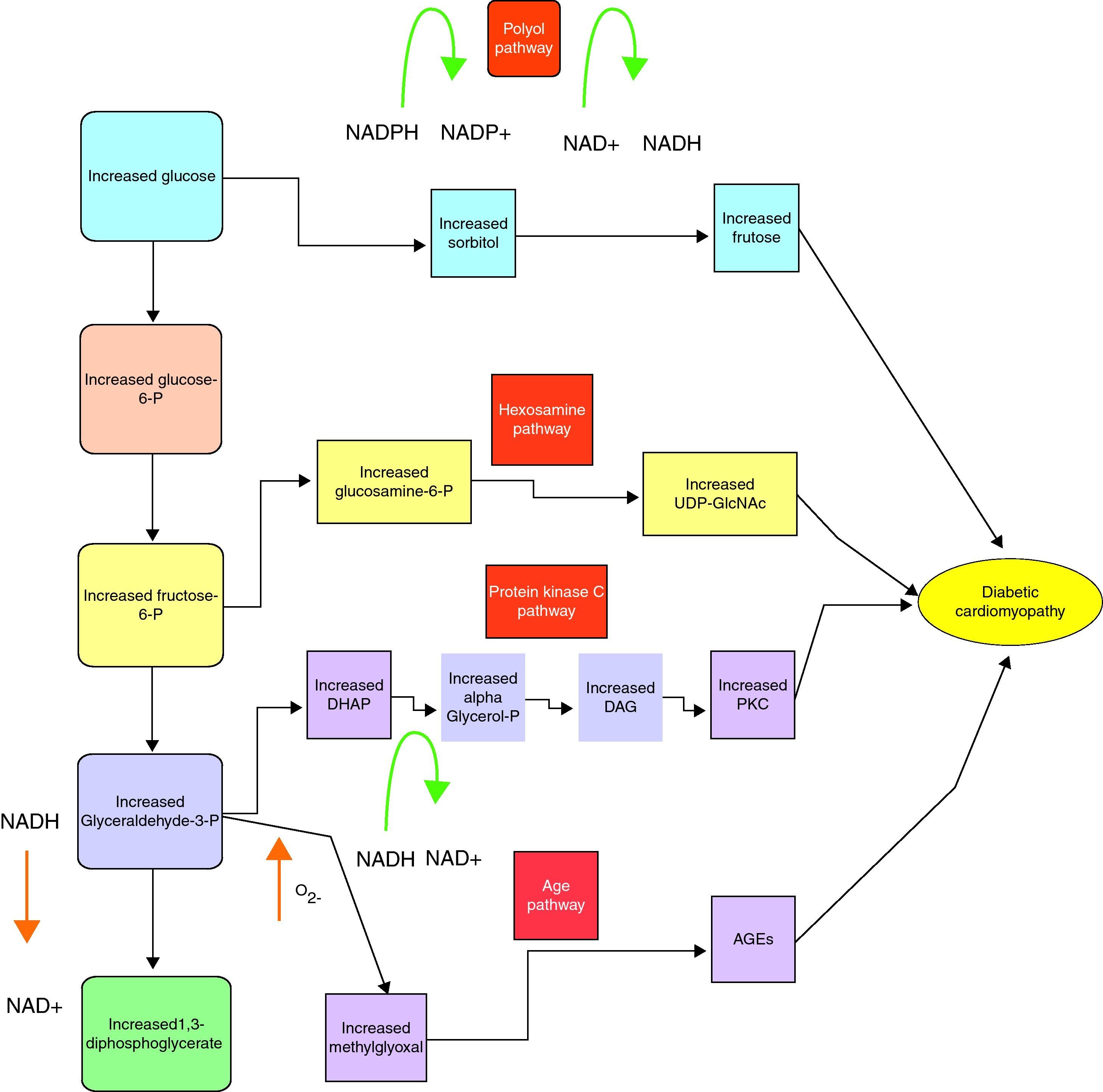
The three main pathological mechanisms by which reactive oxygen species (ROS) are created in diabetes are hyperglycaemia, increased fatty acids and fatty acid oxidation (outlined earlier) and mitochondrial uncoupling.14
The hypothesised first step in the hyperglycaemia pathway is excess sugar forced into the polyol pathway which converts the NADPH to NADP removing glutathione, a very important antioxidant from the system and thus making a normal level of ROS uncontrollable.15 Hyperglycaemia also upregulates the activity of protein kinase C (PKC), which while having a plethora of deleterious effects on vessels, up-regulates the pro-inflammatory NF-KB pathway and causes an increase in ROS generation via NADPH oxidase.16 Excess glucose can also be forced into the hexosamine pathway, which increases production of two inflammatory cytokines TGF-beta and plasminogen activator inhibitor type one.17
Hyperglycaemia thus forces excess glucose into pathways, producing harmful effects on the heart. Hyperglycaemia induced ROS creation is not just a consequence of the above pathways but is also factor that promotes the excess glucose to be forced into them.15,18,19
A review of the metabolic pathways linked to hyperglycaemia is shown below20 (fig. 1).

Mitochondria as the key energy creator are vitally important to areas of high-energy usage such as the heart. In diabetic mouse models, increased ROS generation resulted in an increase in mitochondrial uncoupling and impairment of cardiac mitochondrial function, through changes in the genetic expression of proteins involved in mitochondrial respiration.21,22
This means that the diabetic heart simply cannot generate enough energy to perform as needed in normal conditions and under stress
Using the free radical damage model outlined above, we can begin to view cardiomyopathy in diabetes as a problem with cellular repair and renewal. In knockout diabetic mouse models where the P66 (a pro-apoptotic gene) gene had been deleted, there was an improvement in telomere length and in cardiac progenitor cell numbers, and therefore the number of myocytes.23 P66 is predominantly activated by excess untreated ROS in diabetes and again gives credence to ROS generation being central to all of the processes causing damage in diabetic cardiomyopathy. P66 has been shown to be increased in other animal models of heart failure, and may be one of the missing links in terms of explaining why cardiomyopathy related to diabetes carries such a poor prognosis.24 The activation of P66 by ROS is also dependent on angiotensin-II binding and this may explain to a certain degree some of the benefits in heart failure of angiotensin converting enzyme inhibitors, particularly in people with diabetes, above and beyond the haemodynamic benefits.25
Calcium Signalling - The Ryanodine ReceptorCalcium is the principal signalling molecule that activates the contractile machinery of the ventricular myocyte. Calcium release to levels high enough to activate contraction are controlled through release of sarcoplasmic calcium via the ryanodine receptor.26,27
The ryanodine complex is affected directly in diabetic cardiomyopathy and this leads to a reduction in its sensitivity to calcium, a persistent leak leading to a reduction in peak levels of calcium and a reduction in the number of receptors, all of which lead to reduced myocardial contractility.27
It has been shown that in patients with diabetes and with heart failure there is a reduction in the expression of the ryanodine receptor and associated factors such as sarcoplasmic reticulum Ca2-ATPase (SECRCA 2) through down-regulation again of the extremely important MEF2C, which regulates SERCA2 and well as GLUT-4.28
So while receptor numbers are reduced, receptor function is also reduced and thought to be caused by ROS induced damage to the receptor and also through a change in the receptor structure due to advanced glycosylation that render the receptor less sensitive to its ligands.29 Chronic damage in diabetes means that there is often dysynchronous release and a persistent diastolic leak impacting on contractility and increasing arrhythmia rates.30
Cardiac Autonomic Neuropathy (CAN)Long before the pathological mechanisms could be understood, clinicians had noticed that diabetic patients had reduced cardiac autonomic function, as measured using R-R variability.31
Eventually it was realized that diabetes affected neuronal function in the heart and this can be through many of the already discussed metabolic problems, such as ROS derived damage and AGEs.
CAN has been shown to lead to not necessarily reduced coronary blood flow32 but a reduction in coronary blood flow reserve, i.e. the flow under stress leading to regular periods of small but significant ischaemia causing cardiac injury.33 This is thought to be due to an exaggerated sympathetic tone being less responsive to vasodilatory factors.
As the autonomic neuropathy progresses there is over-production of catecholamines and increased beta-adrenergic receptor production in the heart. These two factors combine to cause an increase in calcium related myocyte apoptosis via endonucleases,34 which progressively leads to heart failure.
While CAN has many effects on vessels and myocytes, the neuronal damage also leads to the loss of calcitonin gene related peptide function, which is released directly from nerves. In diabetes this peptide's receptor is down-regulated and the peptide itself is not released from the nerve; this is important as this peptide causes vessel relaxation and increases cardiac contractility.35
Anti-Diuretic HormoneWhat we have seen so far is that there are a wide variety of metabolic and inflammatory changes all damaging the heart and its function, but not necessarily affecting the volume status of the patient.
One of the newest areas of discussion in heart failure surround the antidiuretic hormone (ADH) and we will look at how this may also complicate diabetic cardiomyopathy.
ADH has a wide variety of effects mediated via three receptors, V1aR (widely expressed), V1bR (pituitary and pancreas) and V2R (renal collecting duct).36 Beyond the anti-diuretic effect, ADH mediates powerful prothrombotic and vasoconstrictive effects via the V1a receptor and hypercortisolaemia via the V1b receptor.36
It is not clear how ADH is up-regulated in diabetes and whether it is a result of endothelial damage37 or whether it plays a primary role in the development of diabetes.36 ADH levels may in the future predict those who go on to develop diabetes, and it has been shown that an infusion of ADH also leads to a massive surge in blood glucose levels in healthy people.36
It has been shown through work on knock out rats lacking V1a receptors; that the lack of this receptor causes impaired glucose tolerance, insulin resistance and high ADH. Similarly, impaired signalling, through V1aR38 leads to elevated levels of ADH, which stimulates V1b (which a lack of in mice caused good glycaemic control) to negatively affect glycaemic control.36
There are increased levels of ADH in patients with diabetes, as discussed above, regardless of the presence of heart failure, and there is also an increased level in patients without diabetes with poor outcome heart failure or myocardial infarction.39,40 ADH causes excess fluid retention and a hypercortisolaemic process regardless of the patient's fluid status, changes which are both likely to cause heart failure.36 There have been small-scale critical care studies, which show that at high doses, ADH increases coronary artery vascular resistance and reduces cardiac inotropy. It is not clear whether the pathological levels seen in diabetic heart failure would be enough to replicate these effects.41
These changes may explain why high ADH levels in heart failure has been shown to be a negative predictor of outcomes in patients39 and may in the future be recognised as a key feature of diabetic cardiomyopathy.
So far we have looked at the changes in heart metabolism and now we need to see how these metabolic and cellular changes affect the heart and lead to heart failure.
Cardiac StructureDiabetes and the prolonged effect of hyperglycaemia have metabolic effects on the heart, and some of these as described previously lead to changes in cellular function, but they also produce changes in the structure of the diabetic heart. Endomyocardial biopsies from diabetic patients show thickening of the capillary basement membrane, myocellular atrophy and hypertrophy with interstitial and myocardial fibrosis and collagen deposition, all of which reduce myocardial function.42
The most common structural problems seen are in left ventricular hypertrophy and cardiac fibrosis, which we will now look at in turn.
In diabetes, up-regulation of the protein kinase C (PKC) system produces increased TGF-B and connective tissue growth factor (CTGF) expression leading to fibrosis and consequently scarred poorly relaxing and poorly contractile hearts.43 This increased fibrosis may also be secondary to a variety of elevated circulating proteins, such as angiotensin and endothelin caused by the constantly occurring inflammatory metabolic and cellular changes.44
One of the central pathological mechanisms in diabetes is the formation of advanced glycosylation end products (AGEs). AGEs are a heterogeneous group of molecules formed non-enzymatically when reducing sugars react with the free amino groups of proteins.45 These products cause cross-linking between myocardial collagen fibres leading to a decrease in compliance and heart failure.46 AGEs up-regulate the genetic expression of nuclear factor kappa B, endothelin and fibronectin which have been linked with myocardial fibrosis, inflammatory changes and increased apoptosis.44
This occurs because receptors (Receptor of advanced glycosylation end products (RAGEs)) on the cell surface are activated by the high levels of AGEs and they cause downstream intracellular activation of various transcription factors, such as nuclear factor Kappa B.47 AGEs are also now thought to cause immune complex formation, in a similar fashion to a variety of other autoimmune diseases and then complement activation and inflammation which causes further damage.48
Patients with diabetes commonly have changes in left ventricular hypertrophy and two causal mechanisms have been most widely investigated.
Hyperinsulinaemia acts as a local growth factor causing increased left ventricular mass via insulin receptor substrate-1 (IRS1) and activation of IRS1-associated phosphatidylinositol-3 kinase (PI3K).49
The other postulated mechanism is that obese patients with type 2 diabetes have high interleukin-6 levels50 and high leptin levels51 which, stimulate growth factors that increase left ventricular mass.
ConclusionIn the above sections we have seen all the various pathological strands currently understood in diabetic cardiomyopathy, but it is important to know that these are all occurring simultaneously in the patient with diabetes. These pathological changes are progressive and are what make diabetic cardiomyopathy the clinical and therapeutic challenge it is today.
As our understanding of the specific pathologies improve, this should lead to the discovery of new therapeutic strategies which will prove useful in tackling the wide cardiac pathology caused by diabetes.
Conflict of interestsThe authors declare no conflicts of interest.
