


Insulin resistance and relative insulin deficiency contribute to the pathogenesis of type 2 diabetes. Defective insulin secretion from pancreatic β-cells results from the progressive deterioration of pancreatic β-cell mass and function. Glucagon-like peptide 1 (GLP-1), an incretin hormone secreted by intestinal L cells in response to a meal, improves glycemic control in patients with type 2 diabetes by addressing both the insulin secretion defect as well as the decline in β-cell mass. These observations fostered the development of new therapeutic agents targeting GLP-1 signaling. This review gives an overview our current knowledge of the molecular mechanisms by which GLP-1 enhances β-cell mass and function.
La resistencia a la insulina y la deficiencia relativa de insulina contribuyen a la patogénesis de la diabetes mellitus tipo 2. La secreción defectuosa de insulina de las células pancreáticas β resulta del deterioro progresivo de la masa y la función de las células pancreáticas β. El péptido similar al glucagón 1 (GLP-1), una hormona incretina secretada por las células intestinales L en respuesta a la ingesta de comida, mejora el control glucémico en pacientes con diabetes mellitus tipo 2 abordando ambos, la deficiente secreción de la insulina, así como el decline de la masa en células β. Estas observaciones fomentan el desarrollo de nuevos agentes terapéuticos orientados a la señalización de GLP-1. Esta reseña resume nuestro actual conocimiento de los mecanismos moleculares por los cuales GLP-1 mejora la masa y función de las células β.
The incidence of type 2 diabetes, a metabolic disorder often associated with obesity, is increasing at an alarming rate1,2. Type 2 diabetes results from the combination of both insulin resistance and progressive deterioration of β-cell mass and function3. In insulin resistant states, β-cells initially compensate for the increased physiological demand for insulin by increasing insulin secretion to efficiently maintain normoglycemia4. Long-standing evidence gathered from rodent models and autopsies in humans indicates that, during this initial stage, β-cell mass is also increased. This may represent an adaptive mechanism to help cope with the metabolic burden. However, β-cell compensation is transient and β-cell mass/function eventually declines in some individuals, thereby causing impaired glucose tolerance or impaired fasting glucose, two hallmarks of prediabetic states. It is hypothesized that the resulting postprandial hyperglycemic episodes may accelerate β-cell demise, a concept called «glucotoxicity»5. When β-cell mass reaches a critical threshold and normal glucose levels can no longer be maintained, type 2 diabetes develops. Indeed, morphometric analyses of pancreases from cadaveric donors demonstrate that β-cell mass is reduced by >50% in individuals with type 2 diabetes compared to control subjects6. Altogether, these observations illustrate the pivotal role of β-cells in the etiology of diabetes. New approaches for diabetes treatment should aim at the preservation and the enhancement of pancreatic β-cell mass/function7.
The incretin hormone GLP-1: an anti-diabetes medicationGlucagon-like peptide-1 (GLP-1) is an incretin hormone secreted by intestinal L cells in response to a meal8. The incretin effect refers to the greater insulin response observed after an oral glucose load compared to a comparable intravenous glucose challenge9. It is estimated that the incretin effect could account for up to 60% of the insulin secretory response in healthy subjects10. Importantly, the incretin response is hampered in patients with type 2 diabetes. This could be attributed to reduced GLP-1 secretion11 and/or GLP-1 receptor (GLP-1R) expression12. The observation that GLP-1 treatment can restore glycemic control in patients with type 2 diabetes13 has rapidly fostered the development of new therapeutic agents targeting GLP-1 signaling for diabetes treatment.
The rapid degradation of native GLP-1 by dipeptidyl peptidase 4 (DPP4)14 represents a limitation to its use as an anti-diabetes medication. Thus, long-lasting GLP-1 analogs that are resistant to the action of DPP4 were developed and characterized as a way to circumvent this major obstacle. The most notorious example is Exendin4, a GLP-1R agonist isolated from the salivary glands of the venomous Gila monster lizard. Another promising therapeutic approach is to prolong the physiological action of endogenous GLP-1 using small molecule inhibitors of DPP4. Because several of these compounds are in various stages of development or already on the market, GLP-1 mimetics and enhancers represent a novel class of antidiabetes medications with a major impact in the treatment of type 2 diabetes mellitus15.
Anti-diabetic actions of GLP-1GLP-1 exerts numerous beneficial effects that improve glycemic control in diabetic subjects. As mentioned above, the stimulation of β-cell insulin secretion is a prominent action of GLP-116,17. GLP-1 also stimulates insulin gene expression and insulin biosynthesis18, at least in part via increased expression and activity of the β-cell specific transcription factor pancreatic and duodenal homeobox gene-1 (Pdx1 )19,20; restores glucose competence in nonresponsive β-cells21; and promotes β-cell mass expansion by stimulating cellular proliferation, survival and differentiation. Thus, GLP-1 addresses both the decline in β-cell mass and the deterioration of β-cell function, two defects that contribute to the etiology of type 2 diabetes. Moreover, GLP-1 has been shown to diminish glucagon secretion13. Noteworthy, the actions of GLP-1 on insulin and glucagon secretion are glucose-dependent, thus only occurring at elevated glucose concentrations. This considerably lowers the risk for hypoglycemia and represents a great advantage over other diabetes medications. Notable extra-pancreatic actions of GLP-1 include8: delay of gastric emptying, which slows the absorption of glucose and nutrients from the gut; inhibition of food intake, which promotes weight loss; and an insulinmimetic action in peripheral tissues, although the presence of an active GLP-1R remains to be demonstrated in peripheral tissues.
GLP-1RGLP-1R is a G-protein-coupled receptor (GPCR) of the B-class subfamily22. It was initially cloned from rat pancreatic islet cells23 and subsequently from a human pancreatic islet library24. The rat and human GLP-1R show a 95% amino acid homology. GLP-1R is coupled to Gs and activates adenylate cylcase to stimulate cAMP production. Canonical downstream effectors of cAMP include protein kinase A (PKA) and cAMPregulated guanine nucleotide exchange factors of the Epac family. In β-cells, GLP-1 triggers Ca2+ signaling and insulin secretion via both PKA and Epac25–27.
Despite several attempts to precisely define the expression pattern of GLP-1R, its tissue distribution remains debated. In pancreatic islets, GLP-1R expression has been shown to be restricted to β-cells in some studies28,29 and to be present in all islet types by others30. Similarly, GLP-1R expression in duct cells is also controversial. The conflicting results could be due to differences in the experimental approaches employed to study GLP-1R expression (western blot, immunohistochemistry, in situ hybridization, and radiolabeled ligand), the specificity/affinity of different GLP-1R antibodies, or variations in GLP-1R expression between species. Besides in pancreas, GLP-1R is found in a small selection of tissues including brain, lung, kidney, heart, and digestive tract28,29.
GLP-1 signaling and insulin secretionGLP-1 stimulates insulin secretion at elevated glucose concentrations that are physiologically observed in the postprandial state (>5mM)21. This unique property increases its therapeutic value since it prevents hypoglycemia, a side effect of other blood glucose lowering agents. Although the precise mechanism is not fully elucidated, the glucoincretin action of GLP-1 has been studied extensively. It was established that GLP-1 facilitates glucose-dependent mitochondrial ATP production31 and promotes the opening of voltage-dependent Ca2+ channels21,27. GLP-1 also increases cAMP levels32, an effect linked to the mobilization of intracellular Ca2+ stores via both the cAMP-binding protein Epac36,33 and PKA31.
The glucoincretin action of GLP-1 is well complemented by its ability to increase insulin biosynthesis18. GLP-1 stimulates Insulin gene expression via increased expression and activity of the transcription factor Pdx1 19,20. Other transcription factors such as NFAT34 and CREB35 could also participate in the process. Increased insulin synthesis and storage is thought to enhance the potential for secretion since it helps maintaining a significant pool of insulin available for exocytosis.
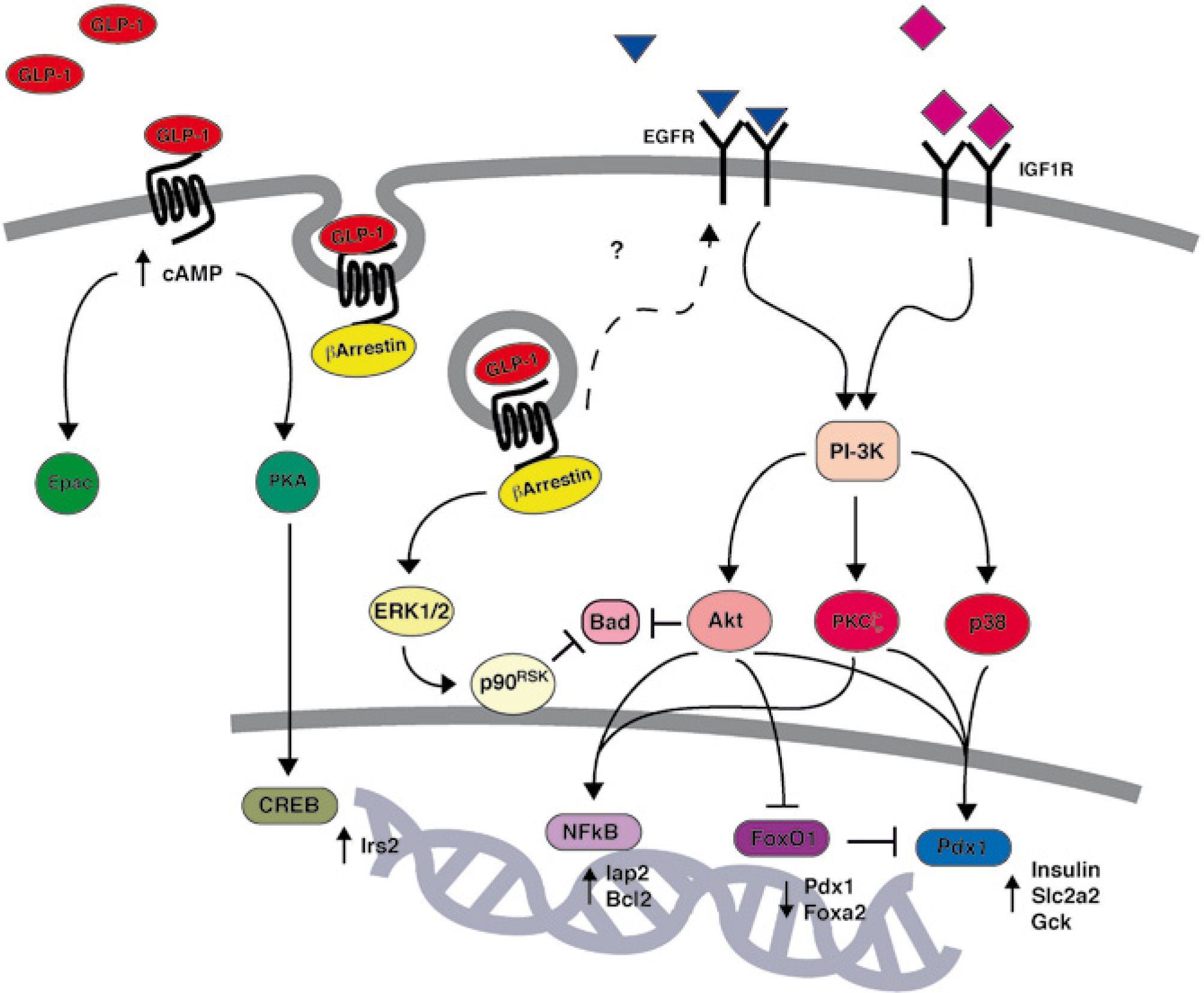
GLP-1 signal transduction and β-cell proliferationGLP-1 acts as a growth factor for the pancreatic β-cell by promoting cellular proliferation and survival. GLP-1 has been initially shown to promote β-cell replication in vitro20,36,37 as well as in vivo in a partial pancreatectomy rat model of type 2 diabetes19. The molecular mechanisms by which GLP-1 promotes β-cell mass expansion have been studied extensively (fig. 1). It was shown that the action of GLP-1 on β-cell mass requires proteolytic maturation of betacellulin by membranebound metalloproteinases37. The release of betacellulin, a member of the EGF family, induces transactivation of the epidermal growth factor receptor (EGFR)37, subsequent activation of PI3K signaling20 and the concerted action of its downstream effectors Akt38–40, PKCζ36, and p38 MAPK36.

GLP-1 activation of PI3K/Akt signaling has been shown to regulate two prominent β-cell transcription factors: Pdx1 and FoxO1. On the one hand, as mentioned previously, GLP-1 increases both the expression and activity of Pdx120. GLP-1-induced activation of Pdx1 is essential for its proliferative and anti-apoptotic effects41. On the other hand, GLP-1 inhibits the Forkhead transcription factor FoxO1 via nuclear exclusion42. Inhibition of FoxO1 by GLP-1 relieves a constraint on β-cell mass expansion as well as Pdx1 expression42. Thus, the Pdx1/FoxO1 tandem unequivocally plays a pivotal role in GLP-1 action.
GLP-1 signaling and β-cell apoptosisThe action of GLP-1 on cellular proliferation dovetails with its effect on survival. Indeed, GLP-1 and its analogs have been shown to prevent β-cell apoptosis in vivo in several rodent models as well as in vitro in response to a variety of environmental stresses. Exendin4 has been shown to delay the onset of diabetes in db/db mice via attenuation of β-cell apoptosis and, consequently, preservation of β-cell mass43. Infusion of native GLP-1 in Zucker diabetic rats promotes β-cell proliferation and prevents apoptosis via caspase-3 inhibition44. Mice with disruption of the Glp1r gene exhibit enhanced β-cell death and more severe hyperglycemia following administration of the β-cell toxin, streptozotocin45.
The precise mechanism by which GLP-1 exerts its antiapoptotic action has been shown to implicate both cAMP/PKA as well as PI3K signaling pathways46. Thus, GLP-1 has been shown to rapidly increase production of the second messenger cAMP to induce PKA-mediated activation of the transcription factor CREB. In turn, CREB enhances expression of Irs2, which acts as a survival factor in β-cells47,48. GLP-1 has also been shown to prevent glucotoxicity as well as lipotoxicity in freshly isolated human islets via PI3K/Akt signaling38. This observation is of great clinical importance since it suggests that GLP-1 could protect β-cells from hyperglycemia and dyslipidemia, two abnormalities that contribute to the development of type 2 diabetes. The proposed mechanism implicates Akt-dependent activation of NF-kB and up-regulation of the anti-apoptotic genes Iap2 and Bcl238. Whereas acute activation of Akt could involve EGFR transactivation37, a new study suggests that prolonged stimulation of Akt could be triggered by an autocrine loop implicating insulin growth factor-2 (IGF-2) secretion and insulin growth factor-1 receptor (IGF-1R) activation49. A recently published study by the Dalle laboratory demonstrated that GLP-1 could also prevent glucotoxicity via barrestin1-ERK1/2-p90RSK-dependent phosphorylation of Bad50. In this model, barrestin1 serves as a docking molecule and creates a signaling complex that favors the sustained activation of ERK1/2. GLP-1 has also been shown to protect β-cells from cytokine-induced cell death41, thereby suggesting a potential therapeutic value of GLP-1 in the treatment of type 1 diabetes. Finally, GLP-1 has been reported to attenuate endoplasmic reticulum (ER) stress in β-cells51,52. ER stress, also called unfolded protein response, designates a cellular stress response related to the endoplasmic reticulum. The underlying mechanisms52 are subject to extensive research and have attracted much interest since the original observation by Yusta et al51 that diabetes is associated with the development of ER stress in β-cells and that GLP-1 signaling prevents ER stress.
ConclusionGLP-1 analogs and enhancers, through their complementary ability to restore and preserve functional β-cell mass, have great therapeutic value in the treatment of type 2 diabetes. Despite being the subject of extensive studies, several questions remain about the biology of incretin hormones. What are the causes of the diminished incretin effect in patients with type 2 diabetes?53. What are the physiological actions of GLP-1 metabolites generated by DPP-4 cleavage of intact GLP-1?54. Furthermore, the elucidation of the precise molecular mechanisms by which GLP-1 enhance β-cell mass and function could lead to the design of new therapeutic agents in diabetes treatment. We can predict that this sizzling field of investigation will continue to keep us entertained in years to come.
Conflict of interestThe authors have no conflict of interest to declare.
JB is supported by a FRSQ junior investigator award and a CDA scholar award.
