


The possibility exists to adopt insulin reduction for preventive and therapeutic purposes in breast cancer. In this regard, recent interest has been focused on the insulin sensitizer metformin, a biguanide derivative that significantly reduces breast cancer incidence and improves breast cancer patient's survival in type 2 diabetics. The ability of metformin to activate AMP-activated protein kinase (AMPK), a key regulator of energy balance in the single cell and the whole organism, largely explains metformin's anti-breast cancer activity. Here, we review the multifaceted and redundant mechanisms through which metformin-reprogrammed energy metabolism at both the organismal and the cellular level may constitute a novel and valuable strategy to prevent and treat breast cancer disease.
acetyl-CoA carboxylase
AMP-activated protein kinase
body mass index
breast cancer susceptibility gene 1
cyclin-dependent kinase
cytokeratin
cAMP-responsive element binding protein
CREB-regulated transcription coactivator 2
disease-free survival
epidermal growth factor receptor
estrogen receptor
fatty acid synthase
fluorouracil/epirubicin/cyclophosphamide
fluorescence in situ hybridization
human epidermal growth factor receptor-2
homeostasis model assessment
gene ontology
insulin growth factor binding proteins
insulin receptor
insulin-like growth factor-receptor 1
serine/ threonine kinase 11 (STK11)
left ventricular ejection fraction
mitogen-activated protein kinase
mammalian target of rapamycin
nicotinamide adenine dinucleotide phosphate
poly(ADP-ribose) polymerase
pathological complete response
prostaglandin E2
progesterone receptor
sterol regulatory element binding protein-1c
tyrosine kinase inhibitor
tuberous sclerosis complex J4
vascular endothelial growth factor
vascular endothelial growth factor receptor
The concept of a relationship between deregulated metabolism and carcinogenesis was first enunciated by Otto Warburg more than 80 years ago.1 Warburg's observation that tumor cells, unlike their normal counterparts, utilize glycolysis instead of mitochondrial oxidative phosphorylation for energy production even when oxygen is present (i.e., the "Warburg effect")2,3 is an old observation that has attracted renewed interest in the last decade.4–7 Previously, changes in cell metabolism that accompanied the malignant phenotype were largely considered a consequence of cellular transformation. Interestingly, the convergence of molecular biology and biochemistry has refocused recent interest in cancer metabolism. Now, altered energy metabolism in tumor cells is regarded as a necessary (and sometimes sufficient) molecular hallmark intrinsically linked to the development and maintenance of the malignant phenotype.8–11
The central role during carcinogenesis that changes in energy metabolism play in preferentially increasing the amount of energy available in tumor cells provides the rationale for hypothesizing that limiting energy availability, that is, the cellular concentration of ATP, could inhibit the carcinogenic process. This notion becomes more apparent when considering that, indeed, other essential hallmarks of cancer disease (e.g., uncontrolled proliferation) are intertwined with an altered tumor cell-intrinsic metabolism, either as a consequence or as cause. Furthermore, there is now a body of evidence that supports a link between obesity, the metabolic syndrome, and insulin resistance with increased risk of several cancers including those of colon and, especially, breast cancer. Hyperinsulinemia, by reflecting the presence of Insulin Resistance Syndrome (IRS) -a condition associated with obesity and physical inactivity that increases risk of diabetes- may represent the molecular link between obesity, other metabolic aspects related to Western lifestyle, and breast cancer risk/metastatic progression. In this regard, since insulin and its related growth factors are widely believed to be mitogenic in an important sub-group of cancer patients, and because pre-operational insulinemia associates with breast cancer progression rates and risk of death, there is a great interest in exploring the possibility that antidiabetic therapies lowering insulin levels could decrease breast cancer incidence and breast cancer-related mortality.
Diabetes, hyperinsulinemia, insulin resistance and breast cancer: identifying targetable molecular connectionsOver the last 10 to 15 years, a substantial body of knowledge has developed regarding the role of insulin and other members of the insulin-like growth factor (IGF) family (i.e., insulin, IGF-1, IGF-2 and, at least, four receptors [IGFR] and six binding proteins [IGFBP]) in breast cáncer.12,13 Although much of the research involving this family has focused on IGFs, notably IGF-1, there has been increasing evidence that physiological concentrations of insulin may play also a clinically important role in breast cancer development. Earlier studies have identified increased risk of breast cancer in women with high insulin levels and, to a lesser extent, in women with type 2 diabetes;14 this is in accordance with the hypothesis that increased insulin levels might promote cancer. Perhaps more relevant is the growing evidence that insulin increases the risk of breast cancer recurrence and death. Goodwin et al.15 identified a significant adverse prognostic effect of fasting insulin levels in locoregional breast cancer. Women with insulin levels in the highest quartile had a double risk of breast cancer recurrence and a triple risk of death. Pasanisi et al.16 similarly reported that either higher insulin levels or the presence of IRS significantly associates with breast cancer mortality. In a correlative study associated with the National Cancer Institute of Canada MA.14 study, Pollak et al.17 reported that women with high levels of C-peptide (a breakdown product formed when insulin is cleaved from pro-insulin) had significantly worse breast cancer outcomes. A recent meta-analysis has confirmed an increased risk of breast cancer in women with diabetes.18 In particular, the meta-analysis of 20 studies (5 case–control and 15 cohort studies) revealed that diabetes was related with an increased risk of several tumor types and the combined results of 10 selected trials found that a high level of insulin associates with a 13% and 25% increased risk of breast carcinoma in casecontrol and cohort studies, respectively. Importantly, all the above mentioned studies have shown that hyperinsulinemia occurring in newly diagnosed breast cancer patients strongly associates with obesity, a well-established breast cancer adverse prognostic factor.19,20
Targeting insulin to prevent and/or treat breast cancer: from lifestyle to pharmacological interventionsAlthough the mechanism(s) by which insulin actively influences breast cancer growth is the subject of intense research, it should be noted that insulin receptors (IR) are almost ubiquitously present in human breast carcinomas and their presence is prognostically relevant.21 Therefore, it is likely that physiological levels of insulin can actively influence breast carcinogenesis by activating several proliferative and antiapoptotic events trough its own receptor.22,23 Moreover, a fetal form of the insulin receptor (IR-a) can be commonly found in breast cancer cells to play a key stimulating role during cell growth.24 Nevertheless, the evidence that insulin influences breast carcinogenesis is extremely important because it raises the possibility of adopting insulin reduction for therapeutic and chemopreventive purposes in breast cancer.
Diet and exerciseIt is likely that lifestyle interventions including weight loss and exercise can lower insulin levels in women with breast cancer. In the breast cancer adjuvant setting, the Women's Intervention and Nutrition Study (WINS) has demonstrated a beneficial effect on disease-free breast cancer survival.25 Although this study did not report on insulin levels, this intervention not only lowered dietary fat intake but resulted further in significant weight loss (up to 2.3 kg relative difference between the two study arms), and the beneficial effects of the intervention were greatest in women with the highest baseline body mass index (BMI). Therefore, it is reasonable to suggest that insulin reduction as a result of weight loss could be an important mediating factor underlying the favourable pro-survival effects of the WINS intervention in breast cancer patients. Supporting this notion, the Women's Healthy Eating and Living Study (WHEL), which evaluated a complex dietary intervention that did not lead to weight loss, failed to identify any survival effects on disease-free survival of breast cancer patients.26 The absence of weight loss (and, likely, insulin reduction) may have contributed to the absence of pro-survival effects in that study.
Conversely to earlier exercise studies in breast cancer that failed to identify insulin-lowering effects of exercise,27,28 Ligibel et al.29 have recently demonstrated that a mixed strength and endurance exercise intervention to sedentary, obese breast cancer survivors resulted in significant statistically significant-improvement in insulin sensitivity as empirically calculated using the Homeostasis Model Assessment (HOMA).
Insulin-sensitizing agentsAlthough lifestyle interventions could lower efficiently insulin levels and, therefore, they can be considered for targeted treatments in breast cancer, we should be conscious that implementation of multimodality weight loss and isolated physical activity interventions aimed to significantly disrupt insulin-related signalling pathways and/or energy factories that account for metabolic reprogramming of tumor cells can be costly and challenging for both patients and practitioners.30 It is obvious that many will prefer the effortlessness and perceived confidence of being treated with a drug that targets cancer's Achilles metabolic heel. In this scenario, recent interest has been focused on metformin, a biguanide derivative, currently approved for the treatment of non insulin-dependent diabetes mellitus, and an insulin-sensitizing agent with potent anti-hyperglycemic properties.31–33 Its primary action is to inhibit hepatic glucose production, but is also increases the sensitivity of peripheral tissue to insulin.34 Thanks to these properties, metformin is an orally administered drug widely used to lower blood glucose concentration in patients with type 2 diabetes and metabolic syndrome. Interestingly, metformin's ability to systemically reduce serum glucose and insulin levels may provide for a potential tumoricidal effect due to its inhibitory effects against the crucial bioenergetics supply aberrantly needed by anabolism-addicted breast cancer cells. Goodwin et al.35 recently reported a phase II study revealing that metformin regimens efficiently lower fasting insulin levels in hyperinsulinemia early stage breast cancer patients. Importantly, metformin treatment reduced insulin levels by 22% in non-diabetic breast cancer patients who had completed primary therapy, which is similar to the reductions with exercise reported by Ligibel et al.29
Metformin & breast cancer treatment: putting the brakes on insulin signalingAt present, and due to ever-growing preclinical studies using tumor-derived cultured cancer cells and animal models, the bench-to-clinic scenario for metformin and breast cancer is rapidly evolving.36 To evaluate the anti-cancer effect of different doses of the drug, we and others have analyzed whether exogenous supplementation with this biguanide could alter cell proliferation profiles of human tumor-derived neuroblastoma, prostate, breast, ovary, colon, glioma, melanoma, endometrial and pancreatic cancer cell lines in vitro.37–56 Remarkably, all these studies have confirmed that metformin acts as an efficient tumor cell growth inhibitor rather than an insulin sensitizer. Moreover, these data have been confirmed in vivo. Thus, either i.p. injected or orally administered with water, metformin administration has been shown to induce significant tumor growth inhibition in human xenografts.39,45,57–60
Metformin against breast cancer: from epidemiology to clinical evidenceThe Russian pioneers. 50 years after its launch for the treatment of type 2 diabetes, we are now leaving a renaissance of the potential anticancer value of metformin. However, as magnificently reviewed recently by Dr. Berstein,33 it should be recognized that metformin has long been known to reduce the growth (and perhaps onset) and progression of tumours. The idea that antidiabetic biguanides may be promising as anticancer drugs was pioneeringly developed by Professor Vladimir Dilman in the early 1970s.61–63 Using phenethylbiguanide (phenformin), a chemical cousin of metformin, he and co-authors at the N. N. Petrov Research Institute of Oncology (St. Petersburg, Russia) achieved the so-called "metabolic rehabilitation" in colon and breast cancer patients. In these patients, phenformin-based clinical management induced retardation of relapses and decrease incidence of primary multiple neoplasias. In animal models, phenformin treatment reduced spontaneous tumor incidence by 80% in C3H mice.64 In the early 2000s, Anisimov's experiments at the Petrov Institute revealed that chronic metformin treatment of female transgenic HER-2/neu mice significantly reduced the incidence and size of mammary adenocarcinomas and increased the mean latency of the tumors.65,66
Epidemiological evidence. Epidemiological studies have confirmed that metformin, but not other anti-diabetic drugs, significantly reduces cancer incidence and improves cancer patients' survival in type 2 diabetics. Evans et al.67 originally reported that the risk of subsequent cancer diagnosis (all cancer types, including breast cancer) was reduced in patients with type 2 diabetes who received metformin (with an odds ratio of 0.85 for any metformin exposure versus no metformin exposure). Importantly, the protective anti-cancer effects of metformin increased with greater metformin exposure (measured as total metformin dose prescribed or total duration of metformin use). Bowker et al.68 reported that cancer mortality was lower in patients with diabetes receiving metformin versus sulfonylureas or insulin (hazard ratio, 0.55 to 0.77), but this study did not evaluate diabetic patients who were not receiving any drug therapy. Recently, Landman et al.69 have evaluated the association between metformin use and cancer mortality in a prospectively followed cohort of 1353 patients with type 2 diabetes enrolled during 1998 and 1999 in the ZODIAC-study in the Netherlands. In this group, metformin use associated also with lower cancer mortality when compared to non-metformin use. Although the design by Landman et al.69 cannot be conclusive about causality, their findings support the notion that metformin use exerts a protective effect on cancer-related mortality. A recent cohort study among people with type 2 diabetes has confirmed further that new users of metformin are at low risk of incident cancer.70 Of note, reduction in cancer risk appears to be metformin-specific and does not occur with all oral anti-diabetic drugs. Thus, a recent study has provided evidence that thiazolidinediones had no effect, whereas effects of sulfonylureas (insulin secretagogues) differed according to the specific drug under evaluation. Indeed, glibenclamide was associated with increased cancer risk.71
Clinical evidence. A real possibility that metformin may have beneficial effects on breast cancer outcome has received definitive clinical support in a recent study conducted by Jiralerspong and colleagues at the M. D. Anderson Cancer Center (Houston, Texas).72 In their study, the authors retrospectively evaluated chemotherapy response rates in a group of 2,592 patients, including 157 women with diabetes, treated with neoadjuvant chemotherapy for early stage or locally advanced breast cancer between 1990 and 2007. Although the number of diabetic patients was small (almost 50% of the diabetic patients initially identified in the database were excluded from the analysis for a number of reasons) and identification of diabetic patients came from patient self-reports without providing any rationale underlying the choice of diabetic agents for a particular agent (thus allowing not only misclassification of some patients but potentially intro- ducing further confounding factors into this retrospective analysis), the authors observed that diabetic patients with breast cancer treated with metformin experienced higher pathological complete response (pCR) rates with neoadjuvant chemotherapy that did those treated with other diabetic medications. Diabetic patients treated with metformin experienced a pCR rate of 24%, which was significantly greater than the pCR rate in diabetic women not treated with metformin (8%; p <0.001) and numerically (but not statistically) greater than the pCR rate in women without diabetes (16%; p= 0.10). Importantly, metformin use was a better predictor of pCR than were well-established breast cancer features (e.g., tumor grade, hormone receptor status, and overexpression of HER2 oncogene) and, in multivariate models adjusting for factors such as BMI, stage, tumor grade, hormone receptor status, HER2 overexpression, age, or presence of diabetes, metformin use remained an independent predictor of pCR with an odd ratio of 2.95 (95% CI, 1.07 to 8.07; p= 0.04).
From a molecular perspective, the study by Jiralerspong et al.72 has provided also a possible link between insulin use and the insulin lowering-related anti-cancer effects of metformin. Exploratory analyses of the relationship between insulin use (which was two-fold higher in the nonmetformin group compared with the metformin group; 33% versus 16%) and pCR revealed a statistically significant association between insulin use and lower pCR rates in the nonmetformin group (0% versus 12%; p= 0.05) but not in the metformin group (27% versus 23%), thus suggesting that insulin use could have contributed to the differences in pCR rates in the two diabetic groups. Despite the fact that pCR rate in diabetic patients treated with metformin tripled over the pCR rate observed in diabetic patients not receiving the drug in this study, there were no differences in rates of disease recurrence among the three groups (i.e., women without diabetes, diabetic women treated with metformin and diabetic women no treated with metformin), and both diabetic groups had worse overall survival compared with the nondiabetic group.
Mechanisms of metformin action against breast cancer: the state-of-artGiven the known pharmacokinetics and wide-spread long-term use clinical use of metformin, its potential use for anti-breast cancer regimens deserves further attention. Metformin has multiple physiological and cellular effects, one or more of which presumably mediates its antitumor activity. Thus, metformin treatment is expected to influence breast cancer disease through non-cell autonomous effects of lowering plasma insulin levels, which itself contributes to cancer risk and incidence, but also through cell-autonomous effect on cell growth. We have suggested recently that metformin is a "hybrid" anticancer compound that physically combines the long-lasting effects of antibodies (by persistently lowering levels of blood insulin and glucose at the physiological level)73,74 and the immediate potency of a cancer cell-targeting molecular agent (by pleiotropically disrupting multiple pro-survival/metastatic axis at the cellular level).36
AMPK-related lowering of blood glucose and insulinThere is growing evidence that metformin effects are largely mediated by stimulation of AMP-activated protein kinase (AMPK).75–77 AMPK is involved in energy-dependent sensing/ signaling and functions as a master metabolic checkpoint to maintain and restore energy homeostasis.78–82 AMPK is activated in response to cellular stresses that deplete cellular energy levels and increase the AMP/ATP ratio. Depletion of ATP and increase of AMP levels can be induced at the cellular level by phenomena such as glucose deprivation, hypoxia, oxidative stress, hyperosmotic stress, tissue ischemia and muscle contraction. Upon activation, AMPK signaling stimulates pathways of energy production while inhibiting those of energy use. Thus, for example, AMPK activation leads to suppression of many of the metabolic processes highly dependent on cellular ATP supply, including gluconeogenesis, protein and fatty acid synthesis, and cholesterol synthesis, as well as stimulation of catabolic processes such as glycolysis and fatty acid β-oxidation. By inhibiting AMPK-regulated transcription of key gluconeogenesis genes in the liver and increasing AMPK-related glucose uptake in skeletal muscle, metformin efficiently reduces levels of circulating glucose, increases insulin sensitivity, and reduces the hyperinsulinemia associated with insulin resistance.83
Metformin appears to activate AMPK by at least two mechanisms that are dependent on its upstream kinase, the tumor suppressor LKB1.84–86 First, metformin inhibits complex I of the mitochondrial respiratory chain, which results in the generation of reactive nitrogen species (ONOO–). ONOO– activates PKCζ (protein kinase C-zeta), which in turn, phosphorylates LKB1 at Ser428.87 Phosphorylation of LKB1 at this residue is required for its translocation from the nucleus to the cytoplasm and subsequent AMPK activation in response to metformin. Second, metformininduced impairment of the mitochondrial respiratory chain results in reduced ATP production and increases of intracellular AMP, which also activated AMPK by an LKB1-dependent mechanism. Importantly, this pathway is required for the therapeutic ability of metformin to lower blood glucose levels.88 Accordingly, as metformin has been more widely prescribed for different insulin-related diseases such as the Polycystic Ovary Syndrome (PCOS), polymorphisms in the LKB1 gene have been found in metformin non-responders.89 Also, genetic polymorphisms in the cell surface transporter organic cation transporter 1 (OCT1), which is required for efficient metformin uptake in hepatocytes, have been shown to underlie metformin resistance in some patients with diabetes type 2.90 Interestingly, the OCT1 transporter shows a limited tissue distribution that is consistent with the pattern of AMPK activation in mice treated with metformin.90,91 Although further attention needs to be paid to whether the effects of metformin in mice and in human epidemiological studies are as a result of reduced insulin levels owing to AMPK activation in liver or as a result of AMPK activation in tumor cells, which leads to suppression of their growth (see below), the fact that insulin and its related growth factors (IGF-1) are widely believed to be mitogenic in an important subgroup of breast cancer patients, and because preoperational insulinemia associates with breast cancer progression rates, AMPK-related systemic reduction of serum levels of glucose and insulin-related growth factors should be proposed as a pivotal molecular mechanism of clinical relevance when employing metformin as a therapeutic agent in breast cancer patients. Nevertheless, non-cell autonomous effects of lowering plasma insulin levels and cell-autonomous effects on tumor cell growth downstream of AMPK need not to be mutually exclusive and both are likely to contribute to the anti-breast cancer actions of metformin.
AMPK-related inhibition of tumoral lipogenesisRapidly proliferating cancer cells require higher rates of de novo lipogenesis to support rapid membrane synthesis. Indeed, lipogenesis is an established hallmark of deregulated metabolism and pathogenicity in cancer early and best adapted (i.e., biologically aggressive) breast cancer phenotypes are addicted to lipid metabolism for cell proliferation and survival.92–96 First, overexpression of certain oncogenes triggers redundant signaling cascades to ensuring that all the major enzymes involved in de novo fatty acid synthesis (e.g., acetyl-CoA carboxylase [ACACA] and fatty acid synthase [FASN]), thus facilitating aerobic glycolysis instead of oxidative phosphorylation for energy production (i.e., the Warburg effect, a major molecular hallmark of malignant cells). Second, certain oncogenes establish a positive bidirectional relationship with key lipogenic enzymes including ACACA and FASN, a feed-back mechanism ensuring a rapid sensing and response to changes in the flux of lipogenic substrates (e.g., NADPH and acetyl-CoA) and lipogenesis endproducts (e.g., palmitate).97–100 Therefore, any (down-stream) inhibitory disturbance in the exacerbated lipogenic activity of tumor cells will result in the immediate (up-stream) blockade of the activity and/or expression of several oncoproteins. Metformin-induced activation of AMPK rapidly induces phosphorylation of ACACA leading to its inhibition and thus blocks the formation of malonyl-CoA, the first committed molecule in the pathways of FA synthesis.101 In addition, metformin-induced activation of AMPK inhibits the expression of lipogenic transcription factor SREBP1c,102–104 which in turn leads to suppression of ACACA and FASN proteins expression and, therefore, to the suppression of the lipogenic phenotype in breast cancer cells.105,106 Besides a role in the maintenance and enhancement of proliferation and survival, breast cancer-associated ACACA and FASN activities are involved also in the production of phospholipids partitioning into detergent-resistant cell membrane microdomains (i.e., lipid rafts aggregates).107 Because raft-aggregates are implicated in key cellular processes including signal transduction, intracellular trafficking, cell polarization, and cell migration,108 metformin-blocked AMPK-regulated lipogenesis might negatively influence aggressive behavior of breast cancer cells not only through the blockade of cell growth and proliferation but also by affecting transduction cascades implicated in the major steps of the metastatic process (i.e., cell adhesion, migration, and invasion).109,110 Indeed, a range of chemical inhibitors of ACACA and FASN are being considered for clinical trials in cancer treatment,94,110 and it is plausible that suppression of lipogenesis is an important part of the tumor suppressor function of AMPK-activating drugs including metformin.
AMPK-related inhibition of mammalian target of rapamycin (mTOR)-regulated protein synthesisIn addition to the effects of metformin and AMPK on bona fide metabolic processes (i.e., circulating levels of glucose and insulin and de novo endogenous biogenesis of fatty acids), metformin-induced activation of AMPK results also in rapid inhibition of cancer cell growth due to the blockade of mTOR-regulated protein synthesis. Activated AMPK phosphorylates and stabilizes the protein product of the tuberous sclerosis complex tumor suppressor gene TSC2, which serves as an integrator and transmitter to mTOR –the master regulator of cellular protein synthesis– of various regulatory inputs implicated in cell growth.111–113 TSC2 not only mediates the inhibitory effects of AMPK on mTOR-regulated protein synthesis but it affects further protein translation by integrating several regulatory inputs (e.g. availability of oxygen and growth factors) sensed and transmitted by two of the most commonly deregulated signaling cascades in human breast cancer (i.e., PI3K/PTEN/Akt and Ras/Raf/Erk).114–116 Since activation of the mTOR pathway frequently occurs in breast cancer and correlates with progression and adverse prognosis in patients,117–119 an important mechanism by which metformin may inhibit breast cancer cell growth and proliferation is through inhibition of mTOR-dependent protein translation.
It should be noted, however, that metformin might also inhibit mTOR in cancer cells independently of AMPK in vivo. Preclinical studies in mice have shown that caloric restriction can decrease IGF-1 levels and inhibit mTOR signaling in multiple tissues.120 Importantly, mTOR inhibition occurred in the absence of AMPK activation, but correlated with inhibition of IGF-1, insulin receptors and Akt. Therefore, it cannot be excluded that metformin could inhibit mTOR not only by cellular activation of mTOR but as well as by decreasing levels of serum insulin or IGF-1.121 Moreover, the fact that LKB1 protein expression is absent or decreased in a significant percent of breast cancer cell lines and primary tumors-which correlates with poor prognosis in breast cancer patients119 strongly suggests that inhibition of tumorigenesis by metformin may depend on the status of LKB1 in cancer patients.122,123 Zakikhani et al.40 originally revealed that exposure of cultured breast cancer cell lines to growth inhibitory concentrations of metformin leads to general declines in mTORregulated protein synthesis and ultimately to efficient blockade of breast cancer cell growth and proliferation in an LKB1-dependent manner. Similarly, treatment of embryonic stem cells with metformin results in growth suppression, an effect that is lost in LKB1-deficient embryonic stem cells.91
AMPK-related inhibition of p53-regulated cell cycle and mitosisAMPK activation not only reprogrammes metabolism, but also enforces a metabolic checkpoint on the cell cycle through its effects on mTOR and p53 signaling.123 Accordingly, the ability of metformin to activate AMPK can lead also to a strong and dosedependent suppression of cell proliferation due to inhibitory effects on the cell cycle. By means of up-regulation in the levels of Cyclin D1 protein,39,44 and the consequent inhibition of the correspondent downstream cyclin-dependent kinases (CDKs), metformin-induced activation of AMPK could eventually cause anti-proliferative effects due to G1 cell cycle arrest. Importantly, AMPK connects cell bioenergetics to the p53 pathway –a central regulator of proliferation and survival–. Upon activation of AMPK by many forms of metabolic stress, including glucose starvation, AMPK can induce a cell-cycle arrest by phosphorylating and stabilizing p53.124,125 AMPK-induced p53 activation promotes cellular survival in response to glucose deprivation, and cells that have undergone a p53-dependent metabolic arrest can rapidly enter the cell cycle upon glucose restoration. Persistent activation of AMPK, however, leads to accelerated p53-dependent cellular senescence. The fact that, in response to energetic stress, activation of AMPK induces apoptotic cell death in a p53-dependent manner,126 may underlie the selective toxicity of metformin against p53-deficient cancer cells.58 Because metformin treatment promotes mitochondria impairment through inhibition of complex I of the mitochondrial respiratory chain,127,128 metformin-treated cells compensate for the suppression of oxidative phosphorylation by increasing their rate of glycolysis in a p53-dependent manner.58 Whereas metformin treatment in p53 wild-type cancer cells activates autophagy (a homeostatic catabolic mechanism for intracellular recycling and metabolic regulation that confers stress tolerance, limits damage, and sustains viability under adverse conditions, thus enabling tumor cell survival in stress)129–132, metformin treatment forces a metabolic conversion that p53-deficient cancer cells are unable to execute. p53-deficient cells fail to arrest in response to AMPK activation and continue to proliferate under metformin-mimicked low-energy conditions, leading eventually to cell death. Accordingly, metformin treatment has been shown to preferentially kill isogenic colon cancer xenografts that lacked p53 compared with xenografts that had intact p53 function.58
AMPK sits in between bona flde tumor suppressors in the complex signaling process that connects cell metabolism with cell proliferation (namely the upstream activating AMPK Kinase LKB1 and the downstream effectors p53 and TSC2). This is consistent with the ability of AMPK to promote survival of cells that are faced with metabolic stress that is imposed by oncogenic activation or chronic biophysical constraints.133,134 Therefore, enhanced activity of AMPK render normal cells resistant to oncogenic transformation and to low-oxygen/glucose deprivation stressful conditions found in solid-tumor microenvironments. Although energy stress can promote apoptotic cell death in cells with a defective p53/AMPK/mTOR pathway, cells with an intact AMPK signaling cascade can survival this stimulus. In the latter scenario, treatment with agents that induce energy stress such as metformin could lead to the prolonged survival of tumor cells. As with other targeted therapeutics, AMPK-activating drugs such as metformin are likely to be most useful against tumors of specific genotypes or in combination with other targeted therapeutics. More refined genetically engineered mouse tumor models should definitely clarify if oncogenic phenotypes such as loss of p53 sensitize breast carcinomas to metformin. In addition, it should be noted that AMPK functioning is required necessarily for faithful chromosomal segregation during mitosis.135–140 Indeed, tumorsuppressive properties of AMPK might relate to its ability to coordinate sensing of energy resources and the fundamental biological process of genome division during mitosis and cytokinesis. If AMPK activation causally relates to timely initiation and correct progression of the mitotic/cytokinetic program, mitotic catastrophe (as a cell death mechanism) and polyploidization events (by reducing proliferative potential) might be also viewed as mitosis-related modes by which metformin treatment might efficiently inhibit tumor formation in permissive p53-mutated tumor cells with defective cell cycle checkpoints.140
Metformin-related inhibition of the inflammatory component in breast cancer microenvironmentAn inflammatory component is present in the microenvironment of most neoplastic tissues, including breast carcinomas. Indeed, it is a well-accepted fact that chronic inflammation is a major contributor to cancer development and progression.141,142 Accordingly, elevated levels of various non-specific markers of inflammation in the serum have been shown to be associated with poor survival in several human cancers. Pierce et al.143 recently revealed that raised circulating levels of C-reactive protein and serum amyloid A were associated with a significantly reduced overall survival and a trend toward a reduced disease-free survival. Patients with type 2 diabetes tend to have high concentrations of circulating C-reactive protein, suggesting that inflammation may contribute to the higher risk of breast cancer and worse prognosis of malignant disease in these patients.144,145 Inflammation, along with insulin resistance/ chronic hyperinsulinemia and increased bioavailability of steroid hormones, has been also suggested to represent one of the mechanisms by which obesity induces or promotes tumorigenesis.146
Because there is evidence to suggest that metformin has a positive impact on inflammation and endothelial dysfunction,147,148 it thus follows that an anti-inflammatory action of metformin is a potential candidate for the physiological effects underlying its antitumor activity. However, the current clinical data for such an effect in patients with diabetes or impaired glucose tolerance is conflicting. The largest study involving patients with impaired glucose tolerance –derived from the Diabetes Prevention Program trial– showed a statistically significant decrease in serum C-reactive protein in the metformin-treated group versus the control group.149 Smaller studies have supported these findings showing a decrease in C-reactive protein levels with metformin treatment.150,151 In contrast, an interim analysis of the Hyperinsulinemia: The Outcome of its Metabolic Effects (HOME) trial, which compared insulin with or without metformin in patients with type 2 diabetes, showed no significant changes in circulating C-reactive protein.152 Since several other small studies have provided similar results,153–155 a consistent clinical anti-inflammatory action of metformin, at least on the basis of biomarker assessment, has yet to be established in patients with diabetes or impaired glucose tolerance. Moreover, while glucose itself can be considered a pro-inflammatory stimulus, insulin appears to have context-dependent effects that may be either anti- or pro-inflammatory.156–159 Given that metformin acts physiologically to lower glucose and insulin levels, the final outcome on local and/or systemic changes in inflammatory parameters is difficult to predict and requires further study.
AMPK-related inhibition of local production of estrogen within the breast tissueIn a recent article, Brown et al.160 presented evidence for a novel mechanism whereby AMPK-activating strategies can decrease breast cancer risk in postmenopausal women, namely by the inhibition of aromatase expression within the breast.161 Local estrogen produced within the breast is considered a pivotal mechanism that drives breast cancer formation in postmenopausal women.162 Estrogen formation is catalyzed by the cytochrome P450 enzyme aromatase, which is present in breast adipose tissue and epithelium. Inflammatory factors such as prostaglandin E2 (PGE2) stimulates aromatase expression in breast adipose mesenchymal cells by a pathway involving cyclic AMP and cAMP-responsive element binding protein (CREB), which binds to two CREs on the promoter II of the aromatase gene. Interestingly, the LKB1/AMPK pathway is inhibitory of aromatase expression in human breast adipose stromal cells while the LKB1/ AMPK pathway is itself inhibited by tumor-derived inflammatory factors including PGE2. On the one hand, AMPK directly inhibit the actions of CREB-regulated transcription coactivator 2 (CRTC2) by causing its phosphorylation and sequestration in the cytoplasm.163 On the other hand, by inhibiting AMPK, PGE2 causes nuclear translocation of CRTC2 and its increased binding to aromatase promoter II, thus contributing to an increase in aromatase expression. Because the latter phenomenon is prevented using AMPK-activating drugs such as AICAR, this finding has significant therapeutic implications as activation of the LKB 1/ AMPK pathway would be theoretically accompanied by a breast-specific inhibition of aromatase expression in estrogen-dependent breast carcinomas. In this scenario, metformin-based therapeutic interventions may result in inhibition of aromatase expression and hence reduction of local production of estrogen within the breast cancer tissue itself.
Regulation of aromatase expression in the breast by AMPK and CRTC2 might also provide a metformin-sensitive link between obesity and breast cancer risk. Leptin synthesis and plasma levels increases with obesity, higher leptin levels have been found to significantly associate with an increase in breast cancer,164 and exogenous leptin stimulates aromatase expression in cultured breast cancer cells.165 In contrast, serum levels of adiponectin decrease with increased obesity, epidemiological studies have shown an inverse association between serum adiponectin levels and breast cancer risk,166,167 and exogenous adiponectin inhibits growth of cultured breast cancer cells.168 Interestingly, the adipokines leptin and adiponectin act also in opposing manners to regulate the AMPK pathway in human breast adipose stromal cells.160,161 Leptin treatment is accompanied by a decrease in the activation status of AMPK and increased nuclear translocation of CRTC2, hence resulting in enhanced aromatase expression. Conversely, adiponectin treatment leads to increased phosphorylation (activation) of AMPK, thus preventing CRTC2 from entering the nucleus, and hence inhibiting aromatase expression. When compared to slim breast cancer individuals, obese individuals with breast cancer may have higher levels of aromatase expression and estrogen levels within the adipose compartment in the breast tissue due to their enhanced expression and secretion of leptin. Although it should be clarified whether this phenomenon would be sufficient to promote proliferation of adjacent breast epithelial cells, it is reasonable to suggest that AMPK-activating drugs including metformin may represent novel therapeutic approaches aimed to inhibit CREB-dependent regulation of aromatase, a crucial determinant of breast tumor formation through local production of estrogens in postmenopausal women. Metformin-based therapeutic interventions may offer a previously unrecognized approach for preventing the so-called pandemic of obesity developing into a breast cancer pandemic particularly in elderly women.
Metformin and breast cancer: time for action in clinical trialsWhen considering all the experimental, clinical and epidemiologic evidence we have accumulated in the last few years, it is reasonable to suggest that, to definitely establish the potential of metformin as a new class of antitumor agent, additional chemopreventive, neoadjuvant, and adjuvant trials should assess the effects of metformin-based regimens in breast cancer patients.31,32 The unexpected "going from the bedside back to the bench" of the ever-growing list of experimental studies supporting the anticancer efficacy of metformin –a readily available, inexpensive and generally well tolerated oral medication– could explain that, at the time of writing, and in response to the inquiry "metformin" & "cancer", a search in ClinicalTrials.gov –a service of the US NIH that registers federally and privately supported clinical trials conducted in the US and around the world– yields seven open studies evaluating the efficacy and safety of treating cancer patients with the biguanide metformin.36 Perhaps the most important one is a large-scale phase III trial of metformin in the adjuvant breast cancer setting. This inter-group clinical trial, led by the National Cancer Institute of Canada Clinical Trials Group (MA.32), is being proposed to evaluate the effects of metformin on breast cancer outcomes, including recurrence and death.31 This trial will be powered to identify clinically plausible and important effects (hazard ratio, 0.76) including key correlative studies that will explore whether any beneficial effect is seen across a broad group of breast cancer patients (i.e., consistent with a direct effect of metformin on AMPK) or whether beneficial effects are restricted to women with hyperinsulinemia, to those whose tumors are IR positive, or to those whose insulin levels fall in response to metformin treatment (i.e., consistent with an indirect effect of metformin acting through an insulin-mediated mechanism).
At the European Institute of Oncology in Italy, the Division of Cancer Prevention and Genetics is planning a presurgical randomized, double blind, placebo-controlled phase II biomarker trial in which breast cancer patients not suitable for neoadjuvant therapy will be randomly assigned to either metformin (850 mg twice/daily) or placebo tablets (28±7 days) until surgery to evaluate the real activity of metformin on tumor proliferation (as measured by Ki-67).32 Also in Italy, there are two further on-going randomized controlled clinical trials, highly intertwined, on metformin-based primary prevention of breast cancer.169 First, the Plotina study will evaluate the effect of metformin on breast cancer primary prevention and on primary prevention of cardiovascular diseases, and patients are being randomized to the treatment arm (850 mg twice/daily) or placebo. Second, the Milan Study will follow a similar design (i.e. metformin versus placebo) plus a diet-intervention focus based on the reduction of high caloric/high glycemic index food, an increase in vegetable intake as well as 30 minutes of physical activity per day. With an overall sample size of 16,000 postmenopausal women and a 5-year follow-up period (325 incidents of breast cancer cases have been estimated to occur among the trial participants), the results of these two trials will clarify in a clinical setting the chemopreventive abilities of metformin envisioned in experimental studies.
Metformin and breast cancer: looking ahead to the future Metformin and breast cancer molecular subtypesBreast carcinomas show clinical, histological and molecular diversity. Gene expression profiling and biomarkers studies have repeatedly shown five distinct molecular subtypes: luminal A (estrogen receptor [ER] positive and/or progesterone receptor [ER] positive, human epidermal growth factor receptor 2 [HER2] negative), luminal B (ER positive and/or PR positive, HER2 positive), HER2-overexpressing (ER negative, PR negative, HER2 positive), basal-like (ER negative, PR negative, HER2 negative, cytokeratin (CK) 5/6 positive and/or epidermal growth factor receptor [EGFR] positive) and normal breast-like tumors. Importantly, biological processes associated with breast cancer clinical outcome appear to strongly depend on the molecular breast cancer subtypes.170–173 Although the terms basal-like and triple-negative breast cancer are not synonymous (as a small proportion of basallike tumors as defined by mRNA expression profiling is not triple-negative, and viceversa),174 approximately 70% of "triple-negative" breast cancers (ER negative, PR negative, HER2 negative) express basal-markers, resulting in the triple-negative subtype commonly being used as a surrogate marker for the basal-like subtype. Luminal tumors have been associated with the most favourable prognoses, while HER2-overexpressing and basal-like tumors, or their surrogate triple negative tumors, have been associated with the worst prognoses. Based on the premise that both the outcome and the clinical response to conventional treatment are significantly affected by the breast cancer intrinsic subtype, we recently hypothesized that mechanism(s) of metformin sensitivity/resistance should vary also across different breast cancer subtypes. Interestingly, we and others have begun to accumulate evidence that biologically aggressive breast cancer subtypes (i.e. HER2-positive and basal-like) may represent good-responder groups among metformin-treated breast cancer patients.
HER2-positive breast carcinomas. First, metformin has been shown to inhibit mammary carcinogenesis in genetically at risk female mice bearing the HER2 (wt-rat-neu transgene) oncogene. Treatment with metformin in transgenic HER2 mice delayed spontaneous breast tumor development and the tumors that did arise in the metformin group were smaller in size compared to untreated controls.65,66 Second, exogenous supplementation with graded concentrations of metformin exquisitely suppresses proliferation of HER2-overexpressing human breast cancer cells when compared with HER2-negative breast cancer cells.42 Indeed, metformin treatment appears to mimic the effects of both HER2 tyrosine kinase inhibitors (TKIs) and anti-HER2 monoclonal antibodies. Thus, in HER2-positive cultured breast cancer cells, low therapeutic concentrations of metformin have been found to block HER2 TK activity while suppression of HER2 protein expression can occur when using higher concentrations of the drug.43 In this regard, we recently reported that exogenous supplementation with metformin synergistically interacts with the HER2 TKI lapatinib (Tykerb®) in HER2-positive breast cancer models with acquired auto-resistance to lapatinib.46,48 Fourth, using whole human genome microarrays (i.e. Agilent 44K Whole Human genome Oligo Microarrays containing 45,220 probes representing 41,000 unique human genes and transcripts) we recently assessed metformin-induced global changes in gene expression identified in HER2-negative and HER2-positive human breast cancer cells. Remarkably, Gene Ontology (GO) term enrichment analyses of metformin-regulated gene clusters revealed that mitosis phases of cell cycle and cytoskeleton were the major effector groups negative regulated by metformin in HER2-negative MCF-7 breast cancer cells, whereas functional clusters related to cell death and apoptosis were principally implicated in the action of metformin against MCF-7 cells engineered to overexpress HER2 human gene (MCF-7/HER2 cells)47 In this scenario, metformin's unique mechanism of action inhibiting HER2 activity/expression and blocking mTOR signaling –a well-recognized contributor to resistance of breast cancer cells to several therapies including trastuzumab– may provide for a potential double-strike against the HER2-positive breast tumor itself. On the other hand, metformin lowered levels of circulating insulin and insulin-like growth factor (IGF) can be expected to prevent activation of the IGF-receptor signaling axis, a well-recognized trans-activated signaling pathway causally involved in refractoriness to anti-HER2 therapies.175,176
To avoid overestimation of the potential benefit of experimental metformin-based therapeutic regimens in unselected populations of breast cancer patients, our Division of Clinical Trials at the Catalan Institute of Oncology (ICO; Girona-Spain) has recently decided to couple metformin in a small "proof-of-principle" study with a neoadjuvant regimen including the anti-HER2 therapy trastuzumab as paradigm.177 The population target of this phase II, randomized, open label, multicentric clinical trial of neoadjuvant chemotherapy and trastuzumab with or without metformin in women diagnosed with HER2-positive primary breast cancer (METTEN-01), will be 66 women with histologically confirmed primary HER2-positive (Dako 3+ or FISH+) invasive breast cancer, T2-T4, any N, M0, and suitable for neoadjuvant chemotherapy plus trastuzumab. After biopsy, patients will be randomly assigned to either 12 cycles of weekly paclitaxel (80 mg/m2) followed by four cycles of FEC (Fluorouracil 500 mg/m2/Epirubicin 75 mg/m2/Cyclophosphamide 500 mg/m2) with concomitant weekly trastuzumab and daily metformin (1500 mg per day −500 mg tablet, taken 3 days a day–) for 24 weeks (Arm A; n= 33) or equivalent neoadjuvant chemotherapy with concurrent trastuzumab plus placebo (Arm B; n= 33) for 24 weeks. The primary endpoint will be efficacy in terms of pCR. Secondary end points of the study will include tolerability, 3-year actual disease-free survival (DFS) after surgery, and cardiovascular events other than Left Ventricular Ejection Fraction (LVEF). pCR predictive factors (e.g. tumor proliferation as measured by Ki67 immunohistochemical staining) and assessment of molecular pathways involved in metformin efficacy/resistance (e.g. expression and activation status of several networks and transduction cascades as measured by immunohistochemical assessment of HER-1/-2/-3, IGF1R, AMPK/mTOR/p70S6K1, AKT, MAPK, VEGF/ VEGFR), will be performed by tissue micro arrays analyses using pre- and post-operative biopsy samples.
Triple-negative (basal-like) breast carcinomas. Triple-negative (basal-like) breast cancers are more frequent in specific subgroups of women including pre-menopausal women of color, pre- and post-menopausal women with an elevated waist-to-hip ratio, pre-menopausal women with an increased BMI, women with a family history of breast cancer, and patients with mutational inactivation of the BRCA1 gene.178–180 Indeed, the fact that basal-like breast carcinomas are more frequent in women who are obese or have type 2 diabetes has lead to the suggestion that public health programs aimed towards achieving a healthy weight might reduce the number of poor prognostic triple negative tumors among all breast cancer cases, especially the high-risk African American group.180 Interestingly, Liu et al.45 have recently reported that metformin exhibits unique biological and molecular effects against triple-negative breast cancer cells. First, triple-negative breast cancer cell lines appeared to be more sensitive to metformin than non-triple-negative cells in proliferation assays. Metformin treatment induced apoptosis via both intrinsic and extrinsic caspase signaling cascades and this activation of apoptosis seemed to be unique to triple-negative breast cancer cells. Metformin treatment induced also apoptosis via poly(ADP-ribose) polymerase (PARP) and downregulated signaling through EGFR (including EGFR itself, MAPK and Akt) in a dose- and time-dependent manner. The fact that metformin treatment decreased the incidence and growth of tumors and prolonged survival in nude mice implanted with highly-metastastic basal-like MDA-MB-231 cells strongly supports the translational value of these findings. For instance, clinical exploitation of the unique anti-cancer activity of metformin against triple-negative breast cancer disease may include a combination of metformin and PARP inhibitors to induce cell death or the combination of metformin with agents selectively inhibiting EGFR (~60% of basal-like breast cancers are EGFR-positive).181–185 It remains to be elucidated what are the relative contributions of AMPK-related signaling in cancer cells versus lowering of serum insulin/IGF-1 levels in mediating the anti-tumor effects of metformin in in vivo models of triple-negative breast cancer. Also, forthcoming studies should establish whether or not obese patients, who are enriched in the triple-negative subtype of breast carcinomas, will be more sensitive to the anti-tumor effects of metformin due to an exacerbated dependence on hyperinsulinemia.186 Nevertheless, it is reasonable to suggest that metforminbased regimens are likely to provide benefit in women with triple-negative breast cancer whether or not they have metabolic derangements of glucose or fat metabolism. Obviously, these effects may be magnified in women with obesity, type 2 diabetes or metabolic syndrome at a significantly increased risk of for triplenegative breast cancer as well as worse outcome if they develop breast cancer. In this scenario, metformin might even be considered for risk reduction chemopreventive protocols.
Metformin and breast cancer stem cellsFor decades, breast cancer initiation and development has been regarded as a multistep process that is reflected by the progressive genetic alterations that drive the transformation of normal human cells into highly malignant derivatives.187 In recent years, the experimental demonstration of tumor-initiating cells (popularly known as cancer stem cells) in several human tumors including breast cancer supports tumor hierarchy as a fundamental concept in tumor biology. According to the cancer stem cell hypothesis, tumor cells are heterogenous and only the tumor-initiating cells have the ability to proliferate extensively, give rise to differentiated cells and form new tumors. Thus, primary breast tumors contain subpopulations of replenishing stem-like cells with the ability of both self-renew and give rise to phenotypically diverse progeny.188–193 Furthermore, it seems that tumor-initiating cells might be intrinsically resistant to many conventional cancer therapies, which might explain the current limitations of these agents in curing human malignancies. When using conventional chemotherapy, the number of breast cancer cells decreases, but the proportion of tumor-initiating stem cells is higher than before treatment, thus indicating that cytotoxics efficiently kill differentiated breast cancer cells whereas cancer stem cells –by their nature– are intrinsically resistant to the effects of anti-cancer drugs thereby allowing breast cancer regrowth. Similarly to traditional anticancer cytotoxics, many novel molecularly-targeted agents (i.e., monoclonal antibodies, tyrosine kinase inhibitors, etc.) have been also designed to target rapidly proliferating cancer cells so many tumor-initiating cells might also be relatively insensitive to these agents. Therefore, a major challenge now is to discover agents and strategies that target breast cancer and breast cancer relapse at their apparent source (i.e., tumor-initiating cells). Remarkably, the anti-tumor effects of metformin may unexpectedly depend on the cancer cell "compartment" of breast carcinomas. A landmark study by Hirsch et al.60 has recently revealed that tumor-forming, self-renewing cancer stem cells are exquisitely sensitive to metformin. Whereas low doses of metformin (0.1 or 0.3 mmol/L) failed to significantly affect cell viability in the non-stem population of differentiated cancer cells, these low concentrations of metformin selectively killed cancer stem cells (as defined by the CD44+/CD24-/low phenotype). Consistently with the "dandelion hypothesis" (i.e., analogous to the propensity of dandelion roots to regenerate weeds, regrowth of tumors from an intrinsically chemotherapy-resistant subpopulation has been termed the "dandelion hypothesis")187 in which both dividing differentiated breast cancer cells and tumorigenic (stem-like) breast cancer cells must be targeted to prevent relapse, concurrent treatment with metformin and the well-defined cytotoxics such as the anthracycline doxorubicin was found to reduce tumor mass and prevent relapse much more effectively than either drug alone in a xenografts mouse model.60
Breast cancer response to metformin: molecular phenotypes and stem cells frequency. The unexpected ability of metformin treatment to attack just the root of the dandelion may largely explain the ability of standard clinical doses of metformin to significantly enhance the rate of pCR in diabetic breast cancer patients receiving metformin and neoadjuvant chemotherapy.72 In addition, the fact that metformin specifically targets tumor-initiating cells could underlie the exquisite sensitivity of HER2-positive and basal-like breast carcinomas to metformin. It has been suggested that the more aggressive and refractory cancers contain more tumor-initiating cells. Following cancer therapy, if the tumor contains only mature (differentiated) cells, the cancer usually does not recur. However, if a large number of immature (undifferentiated) cells (probably including a large proportion of tumor-initiating cells) are present in the tumor sample, the cancer is likely to return, and further aggressive treatment is warranted. On the basis of the stem-cell model of breast carcinogenesis, ER-negative basal-like breast tumors (and highly-sensitive to metformin) would arise from the most primitive stem cells. The HER2-positive subtype (and sensitive to metformin) might be derived from a stem cell midway along the continuum. The well-differentiated, ER-positive luminal A subtype (and resistant to metformin) would be predicted to arise from the transformation of ER-positive stem cells. Basal-like breast cancer are characterized as resembling stem-like cells, mainly composed of cells expressing the cancer stem cell markers CD44 and cytokeratin 5/6,194 and recent studies also suggest a pivotal role for HER2 in maintaining tumor-initiating cells in breast cancer in addition to its presumed role in bulk tumor cells.195,196 Therefore, it can be established a positive correlation between high frequency of stem cells in the more primitive breast cancer molecular phenotypes (i.e., basal-like and HER2-positive) and higher rates of response to metformin-based treatments. Regardless the relevance of metformin doses toward metformin's molecular target on either breast cancer cell compartment (IGF-1/IGF1-R1, AMPK/mTOR or both –a crucial issue that certainly merits to be addressed in future studies–), these findings strongly suggest that, in combination with conventional therapy or molecularly-targeted agents, metformin co-treatment may provide a successful therapeutic strategy to prevent breast cancer recurrence and improve long-term survival in breast cancer patients.
Metformin and breast cancer prevention: modulating stem cell number & niches. Because the "cancer stem cell hypothesis" posits that cancers, including breast cancer, may arise in tissue stem or progenitor cells, the risk for developing breast cancer may be determined, at least in part, by the number of breast stem/progenitor cells that can serve as targets for malignant transformation.197 The behavior of normal stem cells is tightly regulated by signals that the cells receive from their microenvironmental niches, which are provided by the adjacent cells and/or extracellular matrix components.198 Importantly, a niche not only supports the self-renewal and maintenance of stem cell identity but also controls stem cell number and proliferation. Since cancer could progress if the niche were expanded or altered through genetic or epigenetic means,198 a strict control of the number and proliferation rate of stem cells might be considered a preventative mechanism against cancer. In this regard, stem cell number may be set during some "critical windows" during development, including in utero, adolescence, and pregnancy. IGF-1 should be considered a potential breast stem cell mitogen that plays an important role in regulating breast stem cell number during these developmental windows.199,200 It might be predicted, therefore, that elevated levels of IGF-1 might trigger expansion of breast stem cell pools. Metformin acting systemically to indirectly lower insulin/IGF-1 levels may significantly decrease the number of targets at risk (i.e., breast stem cells) for transformation, thereby providing a previously unrecognized stem cell-related explanation to landmark epidemiological studies which demonstrated lower breast cancer mortality in patients treated with metformin and a metformin dose-dependent decrease in breast cancer incidence in metformin-treated diabetics. Although it is unclear what molecular mechanisms control the maintenance and survival of breast cancer stem cells, findings by Hirsch et al.60 provide also a strong rationale for studying yet to be explored roles for insulin, IGF-1/IGF1-R1 and AMPK/mTOR signaling as metformin-targetable pathways beyond Notch, Wnt and Hedgehog in stem-cell maintenance.
Metformin: getting reset to metabolically fight cancer (a corollary)Multiple studies have shown that type 2 diabetes and obesity are associated with an increased risk of breast cancer. Given that insulin and insulin-related receptors (IR, IGF-1R) are relatively ubiquitous in breast cancer patients of all molecular subtypes, including highly-aggressive HER2-positive and triple-negative (basal-like) breast carcinomas, and because patients with high levels of insulin typically have a poor outcome, insulin and related insulin-induced signaling are increasingly associated with the breast cancer formation, invasive/metastasic progression and resistance to anti-breast cancer therapies. Supporting this notion, insulin analogues promote mammary epithelial cell growth, with both metabolic and mitogenic effects, via the IR and IGF-1R and downstream cascades PI-3K and MAPK.201,202 Therefore, there is sufficient current evidence to justify the evaluation of a variety of interventions that lower insulin levels as targeted treatments in breast cancer. If these interventions (e.g., multimodality weight loss and isolated physical activity) can lead to insulin reductions in the range of 25% in the adjuvant setting and if those reductions reverse the adverse prognostic effects of insulin that have been reported, then one might expect to see a 5% to 6% absolute improvement in 5-year disease-free survival.30,31 Although apparently low, this is a highly clinically significant effect that would be comparable to that of many commonly used adjuvant therapies in breast cancer.
- •
By lowering plasma insulin levels and other mechanisms at molecular level, metformin may reduce development and progression of breast cancer.
- •
The unexpected ability of metformin to eliminate tumor-initiating (breast cancer stem) cells opens a new perspective to explore the clinical efficacy of new regimens combining classical cytotoxic approaches and metformin.
- •
Metformin's anti-breast cancer actions strongly support the notion that metformin-based pharmacological approaches aimed to reprogram metabolic status at both the organismal and cellular levels may represent a novel and valuable strategy to prevent and/or treat breast cancer disease.
The good news is that an old pharmacological approach may notably augment the basic approach of behavioral and diet modifications in helping breast cancer patients (and, perhaps, on healthy individuals at risk of breast cancer) to manage their metabolic status on a daily basis. This drug is metformin, a readily available, inexpensive and generally well tolerated biguanide currently approved for the treatment of non insulin-dependent diabetes mellitus that possesses a significant anti-breast cancer potential.203 Indeed, a recent commentary in the Journal of the National Cancer Institute reported that "some physicians proposed adding metformin to cancer treatment regimens".204 We acknowledge that many additional epidemiological studies are required to determine whether there is a clear tumor suppressor effect of prolonged use of metformin and, if so, whether breast carcinomas that have specific molecular signatures will show the greatest potential response. Nevertheless, the numerous potential benefits that have been reviewed in this article certainly support the notion that metformin-reprogrammed metabolic status at both the organismal and cellular levels may represent a novel and valuable strategy to prevent and/or treat breast cancer disease (figure 1).
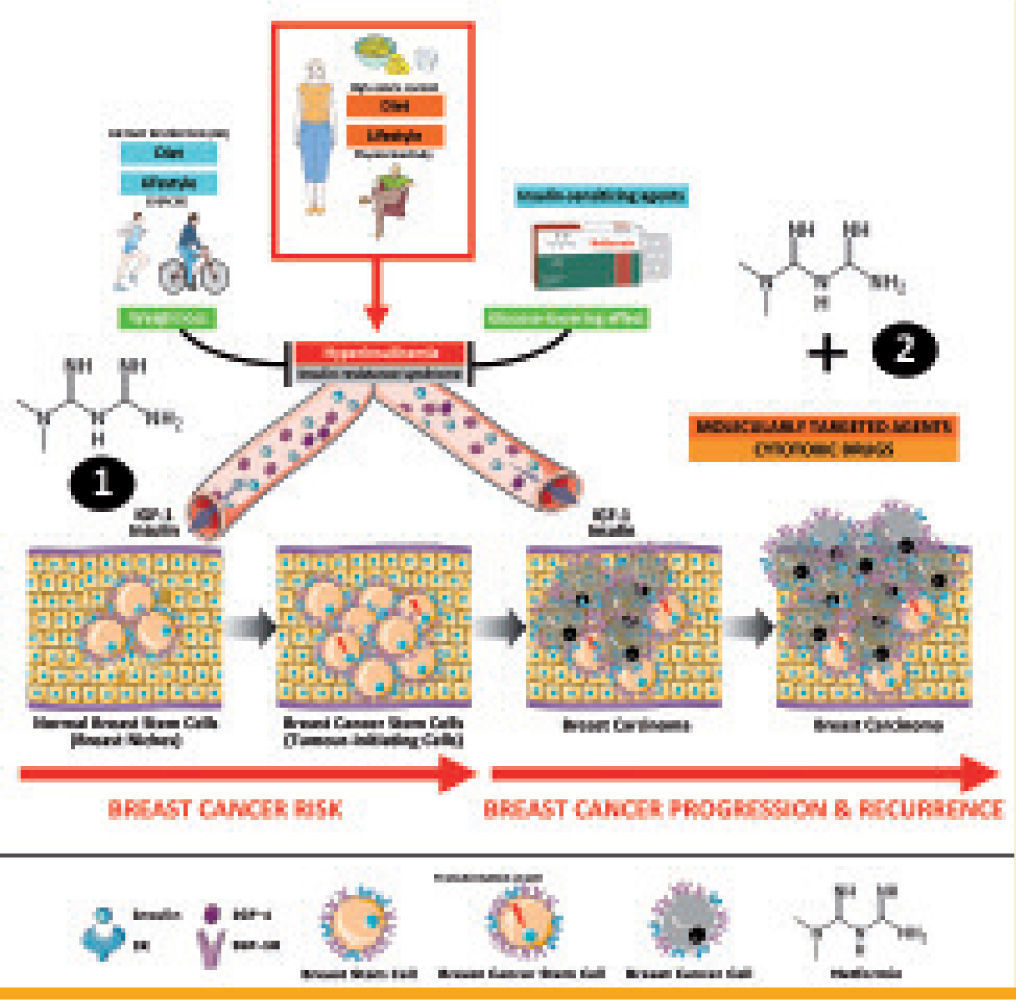
 AMPK activating strategies such as metformin for preventive and therapeutic purposes in breast cancer can be explained through multifaceted and redundant mechanisms involving both tumor-initiating breast cancer stem cells and differentiated breast cancer cells. Insulin and insulin-related growth factors actively increase breast stem cell numbers and/or to maintain breast stem cell niches, thus increasing the number of cell targets at risk of malignant transformation. Owing to
AMPK activating strategies such as metformin for preventive and therapeutic purposes in breast cancer can be explained through multifaceted and redundant mechanisms involving both tumor-initiating breast cancer stem cells and differentiated breast cancer cells. Insulin and insulin-related growth factors actively increase breast stem cell numbers and/or to maintain breast stem cell niches, thus increasing the number of cell targets at risk of malignant transformation. Owing to Energy metabolism and breast cancer disease: a metformin-targetable continuum from breast cancer risk to breast cancer progression and relapse. In accordance with the hypothesis that increased insulin levels might promote BC, hyperinsulinemia –by reflecting the presence of Insulin Resistance Syndrome (IRS)– likely constitutes the causal link to molecularly connect Western lifestyle's metabolic aspects (high-caloric diet- and physical inactivity-related overweight, obesity and diabetes) with an increased risk of BC, disease recurrence and death. The notion of adopting insulin reduction and/or AMPK activating strategies such as metformin for preventive and therapeutic purposes in breast cancer can be explained through multifaceted and redundant mechanisms involving both tumor-initiating breast cancer stem cells and differentiated breast cancer cells. Insulin and insulin-related growth factors actively increase breast stem cell numbers and/or to maintain breast stem cell niches, thus increasing the number of cell targets at risk of malignant transformation. Owing to AMPK activation on the liver (1), metformin-induced inhibition of hepatic gluconeogenesis leads to lowering plasma insulin levels, thus preventing insulin-related signaling in IR/IGF-RI-positive breast cancer cells (i.e., survival and differentiation of tumor-initiating breast cancer stem cells as well as proliferation, survival, migration and invasion of bulk differentiated tumor cells). Metformin treatment, as a result of LKB1- dependent activation of AMPK (2) in breast cancer cells themselves, can efficiently block breast cancer growth and proliferation through multiple mechanisms including tumoral lipogenesis, lipid raft-associated signaling platforms, mTOR-regulated translation initiation and global protein synthesis, Cyclin Dl-regulated cell cycle progression, mitotic division, and p53-regulated autophagy and apoptotic cell death. The unique ability of metformin to target tumor-initiating and bulk differentiated breast cancer cells via non-breast cancer cell autonomous and breast cancer cell autonomous mechanisms may open novel avenues to explore the clinical efficacy of new chemopreventive strategies as well as of new treatment regimens combining classical cytotoxic approaches and/or molecularly-targeted agents with metformin, particularly in hyperinsulinemic women at risk and/or in intrinsically aggressive breast cancer subtypes bearing high number of tumor-initiating stem cells
The authors are not aware of any conflicts of interest related to the subject of the article.
Alejandro Vázquez-Martín is the recipient of a "Sara Borrell" postdoctoral contract (CD08/00283, Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria –FIS–, Spain). This work was supported in part by Instituto de Salud Carlos III (Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria –FIS–, Spain, Grants CP05-00090, PI06-0778 and RD06-0020-0028 to Javier A. Menéndez). Javier A. Menéndez was also supported by a Grant from the Fundación Científica de la Asociación Española Contra el Cáncer (AECC, Spain) and by the Ministerio de Ciencia e Innovación (SAF2009-11579, Plan Nacional de I+D+ I, MICINN, Spain).
