



El proceso de aprobación de nuevos antidiabéticos ha experimentado un cambio radical, que implica a la amplitud, la extensión y los objetivos de su desarrollo clínico. Se han discutido las variables a investigar, el tipo y duración de los ensayos clínicos y lo que debe completarse antes y después de la autorización. Las Agencias del Medicamento norteamericana y Europea han establecido nuevos requisitos con especial énfasis en la seguridad cardiovascular. Los nuevos antidiabéticos deberán mostrar su eficacia sobre el control de la glucemia en monoterapia y/o en tratamiento de combinación, y también un perfil suficiente de seguridad cardiovascular mediante un análisis integrado de eventos cardiovasculares realizado por un Comité Independiente de Adjudicación. En algunos casos será necesario realizar estudios específicos de seguridad con la incidencia de eventos cardiovasculares como variable primaria. El tiempo necesario para el desarrollo clínico de nuevos antidiabéticos podría ampliarse entre 2 y 3 años.
The approval process for new antidiabetic drugs has been radically transformed, involving the scope, extension and objectives of their clinical development. The endpoints, type and duration of the clinical trials, and which ones need to be completed before and after the marketing authorization, have been debated. The US and European Regulatory Agencies have established new requirements for the clinical development of antidiabetic drugs, with special emphasis on cardiovascular safety. New antidiabetic drugs will need proof of efficacy in terms of glycemic control, in monotherapy and/or in combination therapy, as well as sufficient cardiovascular safety, assessed by an integrated analysis of cardiovascular events performed by an independent Event Adjudication Committee. In some cases, specific safety studies with cardiovascular events as primary endpoints may be necessary. The time needed to complete the clinical development of new antidiabetic drugs may be extended by 2-3 years.
El proceso de aprobación de nuevos medicamentos antidiabéticos (AD) ha experimentado un cambio radical, aún en curso de evolución, en los últimos años. No hace mucho que el propio editor de esta publicación se refería al reto que supone escribir sobre nuevos tratamientos hipoglucemiantes1. El debate había comenzado a gestarse con la fuerte crítica efectuada por el reconocido experto David Nathan a la aprobación de un nuevo inhibidor de la dipeptidil peptidasa IV (DPP-IV)2, que fue inmediata y rotundamente contestada desde algunos foros, incluida la propia Food and Drug Administration (FDA) norteamericana3. Con todo, fue el ya archiconocido artículo de Steve Nissen sobre rosiglitazona4 el que sirvió de espoleta para un ulterior debate incendiado hasta límites no habituales en el mundo de la medicina, tanto en lo referente al diluvio mediático de insólito sensacionalismo como a la profusión de artículos de todo tipo en publicaciones médicas de prestigio, cuya sola enumeración excedería con largueza los límites marcados para la bibliografía de este artículo5–8. La crítica no se ha limitado a la industria, sino que ha alcanzado de lleno a las autoridades sanitarias9. La repercusión pública subsiguiente ha tenido un desenlace poco habitual: por una parte, la publicación, tanto por parte de la FDA como de la European Medicines Agency (EMA) de sus posiciones sobre la evaluación y reevaluación del medicamento debatido10,11, y por otra, la revisión, por parte de las mismas agencias, de sus recomendaciones para la aprobación de nuevos AD. La FDA tiene, aún en fase de borrador, una guía general para el desarrollo de AD12, pero se ha centrado fundamentalmente en lo referido a la seguridad cardiovascular13. La EMA, por su parte, cubre, de forma más general, ambos aspectos en su propio borrador14. La presente revisión pretende ofrecer una perspectiva general sobre el complejo panorama al que se enfrenta en estos momentos el desarrollo clínico de nuevos AD.
Las preguntas: necesidades y posibilidadesEn el centro del debate todo un desfile de preguntas, desde las -aparentemente- más obvias y de fácil respuesta a las de más compleja contestación. Las 2 primeras: ¿son eficaces y seguros los AD de que disponemos en la actualidad?, ¿necesitamos más? Si la respuesta a la segunda pregunta es afirmativa, surgen las relacionadas estrictamente con la aprobación técnica por la autoridad sanitaria correspondiente: ¿son suficientes los requisitos, en materia de desarrollo clínico, que las agencias reguladoras exigen a los nuevos AD para su aprobación?, ¿es suficiente la información sobre seguridad disponible en el momento de la autorización?, ¿son suficientemente largos los ensayos clínicos que se llevan a cabo antes de esta?, ¿debe centrarse la evaluación clínica en su acción sobre el metabolismo de la glucosa o en la repercusión cardiovascular? Dentro de esta ¿debe plantearse el asunto como búsqueda de beneficio o como ausencia de riesgo?, ¿se están utilizando en los estudios las variables adecuadas para la valoración de la eficacia?, ¿son aceptables las variables subrogadas o es imprescindible disponer de variables clínicas directas de morbi-mortalidad? Si se utilizan variables de control glucémico, ¿cuál es la adecuada?, ¿es importante la duración del control?, ¿y la protección de la función de la célula beta? Para complicar más la cuestión ¿tiene el control de la glucemia repercusión sobre los eventos cardiovasculares? Por otra parte, tenemos las preguntas que plantean cuestiones de viabilidad: ¿cómo se puede ver afectado el desarrollo de nuevos AD si se demandan grandes estudios de muy larga duración con variables clínicas de índole cardiovascular antes de su aprobación?, ¿es esa una propuesta realmente viable?, ¿existe alguna alternativa?, ¿es suficiente el compromiso de realizar determinados estudios en la fase poscomercialización? Y finalmente, algo que en estos tiempos de crisis, y especialmente en ámbitos de sistemas públicos de salud como el nuestro, no es menos relevante: ¿son todos estos cambios suficientes para satisfacer las expectativas de los responsables del reembolso y financiación de los medicamentos? Analizar y responder todas estas preguntas con detalle es empresa demasiado ambiciosa para el propósito del presente artículo, pero intentaré ofrecer al lector los ingredientes que le permitan hacerse una idea general sobre la complejidad del panorama actual para el desarrollo clínico de estos medicamentos.
¿«Glucocentrismo» … o «cardiocentrismo»?Hay quien ha declarado que ninguno de los antidiabéticos orales disponibles ha probado su eficacia clínica, que enfocar el asunto exclusivamente desde la seguridad cardiovascular y no desde la eficacia clínica es un error, y que la obsesión por el control glucémico es excesiva y probablemente dañina para los pacientes5,15. Desgraciadamente, el advenimiento reciente de estudios grandes y prolongados sobre el impacto del control glucémico en variables clínicas cardiovasculares, bien con tratamientos concretos, bien con estrategias terapéuticas combinadas16–20, lejos de resolver la cuestión, ha dejado una mezcla de confusión y frustración. Estas, que han conducido a conclusiones extremas como las antes citadas, han sido afortunadamente valoradas con buena dosis de equilibrio y sentido común por otros autores21,22. Además, hay también quien ha defendido y sigue defendiendo el valor del control glucémico como variable de eficacia, y el de la hemoglobina glucosilada como parámetro subrogado apropiado12,14. Incluso se ha señalado con razón que hasta el momento ningún AD ha demostrado un efecto favorable sobre las variables cardiovasculares, y apuntado que, sobre dicha base, cabría preguntarse también no solo si los nuevos AD deben aprobarse, sino incluso si los AD «clásicos» deberían permanecer o no disponibles23. Si se intenta centrar el asunto, parece lógico pensar que todos los involucrados en el tratamiento de la diabetes desearíamos, en un escenario ideal, disponer de variables clínicas de las consideradas «duras», como reducción de la mortalidad o de las complicaciones macro o microvasculares severas. También parecería obvio, salvo que busquemos una «epidemia» de cetoacidosis y glucemias incontroladas y despreciemos el documentado beneficio del control glucémico sobre las complicaciones microvasculares24,25, que más allá de los imprescindibles cambios en el estilo de vida, léase dieta y ejercicio, se necesitan medicamentos que proporcionen un control glucémico lo más estable, duradero y seguro posible. Y no parece descabellado considerar que el arsenal terapéutico disponible ha mostrado suficientemente una eficacia al menos razonable en ese sentido, pese a las opiniones contrarias extremas antes mencionadas15. También parece razonable asumir, y así lo hace la FDA26, que con el tiempo, será necesario añadir medicación antidiabética a la inicialmente instaurada, por lo que el tratamiento de combinación, más que una conveniencia, será una necesidad. Y en este sentido, disponer de diferentes posibilidades tampoco parece algo ilógico, aunque esto, por otra parte, complica de forma considerable tanto el diseño como la duración e interpretación de los resultados de los estudios que puedan llevarse a cabo23,26. Por último, es también evidente que la interacción de la diabetes con otros factores de riesgo cardiovascular, y el propio hecho de que la causa cardiovascular está detrás de la mayor morbi-mortalidad de los pacientes diabéticos, hace necesaria la actuación multifactorial integrada26. En este sentido, parece que, una vez más, en el punto medio estaría la virtud, de forma que el ideal quizá fuera disponer de medicamentos que proporcionen un control adecuado del metabolismo de la glucosa, de la forma más fisiológica y favorable para la función de la célula beta, y que favorezcan, o como mínimo no perjudiquen, la función cardiovascular.
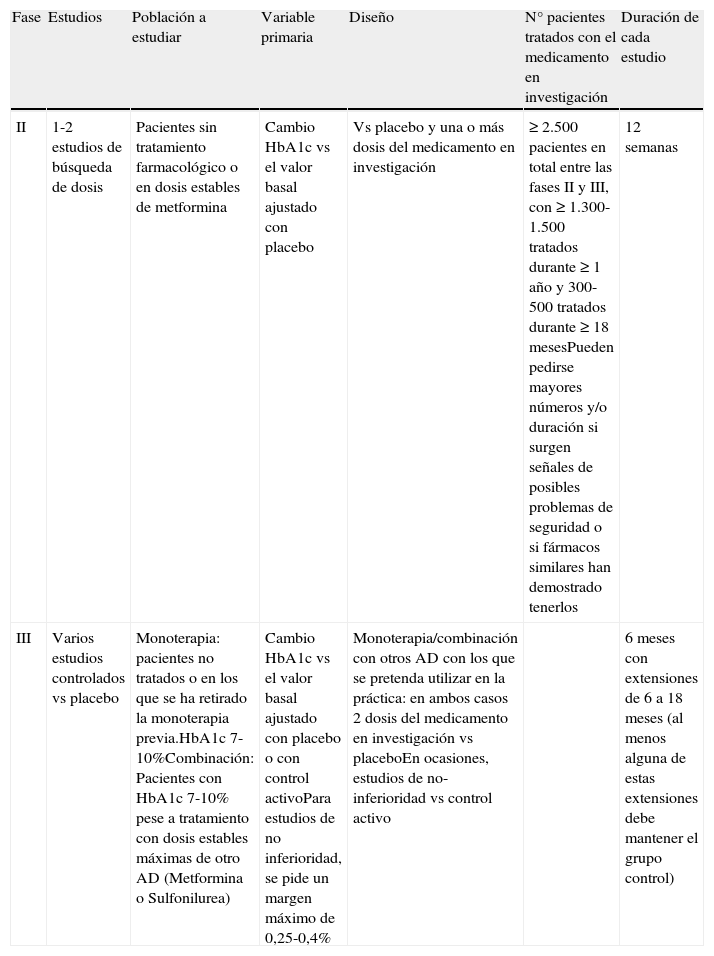
La situación actual: recomendaciones generalesEn el momento actual, y aparte de la evaluación de la seguridad cardiovascular, que repasaré posteriormente, la recomendación general de la FDA para el desarrollo clínico de nuevos medicamentos antidiabéticos aparece resumida en la tabla 1. En Europa, las recomendaciones14 son básicamente parecidas, aunque existen algunas peculiaridades que en lo esencial se resumen en la tabla 2. Hay que resaltar que ambas agencias consideran sus guías como recomendaciones en ningún caso vinculantes, y que ambas recomiendan –y así se hace ahora habitualmente- el asesoramiento científico previo con ellas para que quien desarrolla el medicamento pueda tener una seguridad razonable de que sus planes están en línea con lo que las autoridades esperan. Tanto una como otra Agencia citan las guías generales de eficacia y seguridad de la Conferencia Internacional de Armonización, documentos de consulta obligada para este y otros casos de desarrollo de medicamentos27. Contando con la necesidad de grupos de control con placebo y/o fármaco(s) activo(s), el número total de pacientes contenidos en el documentación presentado para solicitar la autorización de comercialización suele oscilar entre los 4.000 y los 5.000 pacientes. Hay que tener en cuenta, sin embargo, algunos matices. Así, circunstancias como los estudios con controles activos o las señales de potenciales problemas de seguridad observadas en la fase preclínica o clínica, o con medicamentos de la misma familia, pueden hacer aumentar considerablemente el tamaño de la muestra total de pacientes requeridos y/o la duración de los ensayos12. Por otra parte, el manejo adecuado de los aspectos de seguridad, antes e incluso más aún después de la comercialización, es un aspecto esencial y que condiciona de manera evidente el desarrollo clínico de estos –y todos- los medicamentos. Así, es evidente que una estrategia de minimización de riesgos como la vigente actualmente en Europa, que incluye el requisito por parte de la EMA de un plan de minimización y control de riesgos para todos los nuevos medicamentos28, influirá en los estudios que se hagan antes y después de la comercialización. Es importante no olvidar, sin embargo, que por ambicioso y extenso que sea el plan de desarrollo completado antes de la comercialización, la capacidad de detección de determinados eventos infrecuentes es limitada, algo que es bien conocido hace tiempo aplicando una regla estadística sencilla29, pero que a menudo se olvida con demasiada ligereza. Como simple muestra de en qué se traduce dicha regla, baste un ejemplo simple: si exponemos a 5.000 pacientes a un medicamento concreto y no observamos ningún caso de un efecto concreto (llamémosle «A»), podemos descartar, con un 95% de certeza, incidencias para el efecto A del 0,06% o superiores. Si quisiéramos descartar una incidencia igual o superior al 0,03%, deberíamos disponer de 10.000 pacientes expuestos a dicho medicamento.
Resumen de requisitos fundamentales para el desarrollo de antidiabéticos (Borrador de la Guía General de la Food and Drug Administration sobre recomendaciones fundamentales para el desarrollo y aprobación de nuevos antidiabéticos)
| Fase | Estudios | Población a estudiar | Variable primaria | Diseño | N° pacientes tratados con el medicamento en investigación | Duración de cada estudio |
| II | 1-2 estudios de búsqueda de dosis | Pacientes sin tratamiento farmacológico o en dosis estables de metformina | Cambio HbA1c vs el valor basal ajustado con placebo | Vs placebo y una o más dosis del medicamento en investigación | ≥ 2.500 pacientes en total entre las fases II y III, con ≥ 1.300-1.500 tratados durante ≥ 1 año y 300-500 tratados durante ≥ 18 mesesPueden pedirse mayores números y/o duración si surgen señales de posibles problemas de seguridad o si fármacos similares han demostrado tenerlos | 12 semanas |
| III | Varios estudios controlados vs placebo | Monoterapia: pacientes no tratados o en los que se ha retirado la monoterapia previa.HbA1c 7-10%Combinación: Pacientes con HbA1c 7-10% pese a tratamiento con dosis estables máximas de otro AD (Metformina o Sulfonilurea) | Cambio HbA1c vs el valor basal ajustado con placebo o con control activoPara estudios de no inferioridad, se pide un margen máximo de 0,25-0,4% | Monoterapia/combinación con otros AD con los que se pretenda utilizar en la práctica: en ambos casos 2 dosis del medicamento en investigación vs placeboEn ocasiones, estudios de no-inferioridad vs control activo | 6 meses con extensiones de 6 a 18 meses (al menos alguna de estas extensiones debe mantener el grupo control) |
AD: antidiabéticos; HbA1c: hemoglobina glucosilada.
Fuente: cita bibliográfica 12.
Principales elementos específicos en las recomendaciones generales de la European Medicines Agency para el desarrollo de nuevos medicamentos antidiabéticos respecto a las de la Food and Drug Administration
| • Uso del placebo: en estudios de 3-6 meses debe quedar limitado a pacientes con diabetes de corta evolución y HbA1c < 8,5% basal. Pacientes con HbA1c > 10% solo deben incluirse en estudios <3 meses |
| • Necesidad de ajuste de dosis por efecto o hasta dosis máxima tolerada |
| • Justificar la relevancia clínica del cambio conseguido en la HbA1c, incluyendo análisis de respuesta (proporción de pacientes que consiguen o mantienen HbA1c ≤ 7%) |
| • Búsqueda de dosis: se deben explorar al menos 3 dosis. Variable primaria: cambios en la glucemia plasmática; cambios en la HbA1c si el estudio es de más de 8-12 semanas |
| • Estudios de confirmación: debe demostrarse superioridad vs placebo y no inferioridad vs control activo. Duración ≥ 6 meses. Para medicamentos con nuevo mecanismo de acción pueden requerirse estudios controlados de 12 meses |
| • Estudios de combinación: necesaria comparación de la combinación del medicamento nuevo con el establecido vs el establecido solo. El tratamiento de base debe mantenerse estable salvo por motivos de seguridad. Se recomienda estudio controlado vs placebo y metformina (3 grupos). Para este caso, se recomienda solicitar asesoramiento científico a la EMA o a la Agencia Nacional correspondiente. Alternativa: estudio vs metformina que demuestre eficacia superior a metformina |
| • Combinaciones a dosis fijas: si no existen datos de eficacia y seguridad de la combinación por separado (como medicamentos independientes), se pueden requerir estudios en pacientes no respondedores o insuficientemente controlados con las dosis máximas toleradas de la monoterapia de primera línea ya establecida |
| • Combinación con insulina: se recomienda la comparación medicamento nuevo + insulina vs insulina sola vs insulina + metformina |
| • Como regla general, número suficiente de pacientes ancianos o de otras poblaciones especiales (función renal alterada, etc.) para conseguir indicaciones menos restringidas en dichas poblaciones |
EMA: European Medicines Agency; HbA1c: hemoglobina glucosilada Extraído de la cita bibliográfica 14.
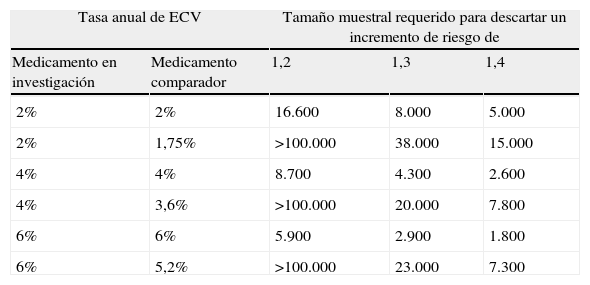
El ejemplo mencionado fue utilizado por la FDA en su reunión con el Comité Asesor de Medicamentos Endocrinológicos y Metabólicos de junio de 2008, dirigida a recoger la opinión de dicho Comité sobre la mejor forma de evaluar la seguridad cardiovascular de los AD30, como oportuno recordatorio sobre lo que se puede detectar o no según los tamaños de muestra utilizados en los ensayos clínicos. En esa densa reunión, cuya transcripción íntegra puede consultarse31, se presentaron también cálculos del número de pacientes que se requeriría en función de la tasa anual de eventos cardiovasculares (ECV) con el medicamento en investigación y con el comparador (tabla 3). El consenso, lógico, de dicha reunión fue que exigir la demostración de beneficio sobre variables cardiovasculares era probablemente una pretensión excesiva, quizá inalcanzable, especialmente teniendo en cuenta que tampoco los AD más «veteranos» entre los comercializados podrían afrontar dicha demanda, como antes señalé. De hecho, la realización de estudios de beneficio cardiovascular en poblaciones con baja tasa anual de ECV exigiría enormes tamaños muestrales y/o seguimientos larguísimos, algo que cabe considerar, por razones obvias, inviable si se exige como paso previo a la autorización de comercialización. La única forma de afrontar este reto con estudios más pequeños y cortos, y por tanto más viables (recordando que, en todo caso hablaríamos de miles de pacientes), sería centrarse en poblaciones de mayor riesgo, que pudieran presentar mayores tasas anuales de ECV. Y aunque la inclusión de dichas poblaciones en los estudios ha sido una de las recomendaciones de las agencias, tal vez procede recordar también que la población con mayor potencial de beneficiarse de un control glucémico adecuado, al menos hasta donde sabemos, es probablemente la que aún no ha desarrollado complicaciones cardiovasculares32. En esa población la tasa anual previsible de eventos cardiovasculares es considerablemente inferior al 2%, situándose probablemente en torno al 0,5%33 o hasta menos, como puede comprobarse en algunos estudios recientes en prediabéticos34. Y estudios con esa tasa anual de ECV en los que pueda documentarse un número suficiente de ellos como para poder descartar con certeza suficiente un exceso de riesgo del 1,3% conllevarían tamaños de cerca de 20.000 pacientes y seguimientos de hasta 5 años35. El panorama se complica aún más si recordamos, como ha señalado la propia FDA, que la mejora en el tratamiento de los factores de riesgo cardiovascular como dislipidemia y otros, redundarán, como ya está ocurriendo, en menores tasas de ECV. Con todos estos ingredientes, cabe considerar la recomendación de la FDA como razonable, aunque es evidente que los nuevos requerimientos añadirán –ya lo están haciendo- no menos de 2-3 años al tiempo necesario (hasta ahora entre 5 y 6 años aproximadamente) para completar el desarrollo clínico de un nuevo AD antes de su comercialización.
Tamaño de los estudios requeridos para la evaluación de seguridad cardiovascular*
| Tasa anual de ECV | Tamaño muestral requerido para descartar un incremento de riesgo de | |||
| Medicamento en investigación | Medicamento comparador | 1,2 | 1,3 | 1,4 |
| 2% | 2% | 16.600 | 8.000 | 5.000 |
| 2% | 1,75% | >100.000 | 38.000 | 15.000 |
| 4% | 4% | 8.700 | 4.300 | 2.600 |
| 4% | 3,6% | >100.000 | 20.000 | 7.800 |
| 6% | 6% | 5.900 | 2.900 | 1.800 |
| 6% | 5,2% | >100.000 | 23.000 | 7.300 |
*En función de: a) la tasa anual de eventos cardiovasculares observada con el medicamento en investigación y con el grupo control, y b) el grado de exceso de riesgo relativo que desea descartarse (ver cita bibliográfica 30).
Los cálculos se basan en un error alfa = 0,05, poder estadístico del 90%, con un seguimiento de 5 años y un periodo de inclusión de pacientes de 2 años.
Extraído de la cita bibliográfica 13.
El asunto de las variables a incluir para la evaluación de la seguridad cardiovascular tiene también importantes implicaciones, cuyo análisis detallado requeriría, por su complejidad36, una revisión detallada aparte más allá de los límites de la presente. La mejora de los tratamientos y la subsiguiente reducción del número de eventos ha convertido en inviable la posibilidad de realizar estudios de tamaño y duración razonables con una sola variable principal, sea esta mortalidad, infarto o cualquier otro ECV importante. La utilización de variables cardiovasculares combinadas deviene así obligada, y de ahí derivan a su vez algunas complejidades, desde cómo definir y valorar determinadas variables hasta su documentación, adjudicación e interpretación37. En primer lugar, hay que definir los componentes de esa variable compuesta. Se ha hecho popular en los estudios cardiovasculares el acrónimo anglosajón MACE («Major Adverse Cardiovascular Events») para designar los ECV importantes. Lo más frecuente es que en esta variable combinada entren los siguientes componentes: mortalidad de cualquier causa (o mortalidad cardiovascular), infarto de miocardio no fatal y accidente cerebrovascular (ACV). Sin embargo, bajo este término genérico pueden incluirse también en determinados casos otras variables, como revascularización coronaria, revascularización de miembro inferior o amputación de miembro inferior38, de forma que MACE puede terminar significando cosas diferentes. Además, la introducción de determinados elementos en esta variable compuesta puede restarle relevancia clínica e incluso, potencialmente, hacer depender el resultado global de dicha variable compuesta del peso excesivo de alguno de sus componentes, a veces de menor impacto clínico, como bien se ha señalado39,40. Y lo que es peor, incluso caben –y se dan- diferentes definiciones para una misma variable. Tal ocurre, por ejemplo, en los casos de mortalidad cardiovascular o en los de hospitalización por causa cardiovascular40. Incluso el componente de infarto no fatal puede tener diferentes consideraciones, dependiendo, por ejemplo, de que en esa categoría se incluyan, o no, los casos de infarto silente. En todo caso, el análisis de la variable MACE, sean cuales fueren los elementos que la compongan, pasa por una definición clara de los mismos en el protocolo, una necesidad de documentar adecuadamente los casos (aspecto esencial sin el que la labor del Comité Independiente de Adjudicación, por competente que este sea, se vuelve imposible) y una monitorización y comunicación adecuada de los mismos41,42. Como se ha comentado durante la mencionada reunión de la FDA31, es esencial eliminar la recogida de datos superfluos pero exigir el máximo de datos fundamentales, como para el caso de un infarto puedan ser el electrocardiograma o los niveles de troponina. Es esencial igualmente asegurar un seguimiento completo y evitar que se pierdan pacientes a efectos de seguimiento13,43. Y es asimismo importante evitar el error, relativamente frecuente, de sobreestimar la incidencia prevista de ECV, que puede dar como resultado un estudio inútil si la incidencia es considerablemente inferior a la prevista. Definir a priori un número global de ECV requerido para que el análisis estadístico sea viable (diseño «event-driven») o alguna otra estrategia adaptativa44 es un requisito, hoy por hoy, casi inexcusable.
Las recomendaciones de las Agencias ReguladorasLas recomendaciones finales de la FDA13 solicitan para el desarrollo clínico de nuevos AD las siguientes acciones en lo referido a la evaluación cardiovascular: a) establecer un Comité Independiente de Adjudicación de ECV que examine y adjudique todos los ECV observados en los estudios de fase II y III; b) los ECV a incluir son: mortalidad cardiovascular, infarto de miocardio y ACV, pero pueden introducirse además otros como por ejemplo hospitalización por síndrome coronario agudo o procedimientos urgentes de revascularización; c) los ensayos clínicos de fase II y III deben estar diseñados de forma que una vez completados pueda llevarse a cabo un metaanálisis sobre seguridad cardiovascular; d) debe proporcionarse a la agencia un protocolo con detalle estadístico del metaanálisis propuesto y los ECV a analizar. La FDA destaca que, con el fin de evaluar estos aspectos, los ensayos controlados pueden requerir mayores duraciones que las habituales, de manera que pueda valorarse el riesgo cardiovascular a un plazo mínimo de 2 años. El fin del metaanálisis, que debe realizarse antes de la aprobación es definir si se necesitan más estudios sobre seguridad cardiovascular en ese momento, si es posible autorizar el medicamento y proceder con estudios adicionales de seguridad cardiovascular tras la comercialización o si se considera que no es necesario realizar ningún estudio adicional sobre estos aspectos. El metaanálisis debe demostrar que la diferencia en el riesgo relativo de incidencia de ECV entre el medicamento en investigación y el grupo control tiene un límite superior para el intervalo de confianza del 95% (IC95%) de 2 colas inferior a 1,8. Si el metaanálisis de los estudios de fase II y III no es suficiente para demostrar eso, se llevará a cabo un estudio específico de seguridad de suficiente tamaño para que, solo o integrado en el metaanálisis descrito anteriormente, pueda demostrarse el requerimiento citado. En cualquier caso, y además del límite superior del IC95%, debe tenerse en cuenta el estimado de riesgo relativo observado. Así, no se considera aceptable un riesgo relativo de 1,5 incluso si el límite superior del IC95% es inferior a 1,8. Si dicho límite se encuentra entre 1,3 y 1,8, y siempre que el análisis general del balance beneficio-riesgo sea positivo, se requerirá un estudio específico de seguridad cardiovascular a realizar tras la comercialización con el fin de demostrar de forma concluyente que el límite superior del IC95% es inferior a 1,3. El estudio poscomercialización puede servir para cumplir este requisito por sí mismo o combinado en un análisis integrado con los estudios precomercialización. Finalmente, la FDA considera que si el límite superior del IC95% para el riesgo relativo es inferior a 1,3 en el metaanálisis de los estudios de fase II y III, y si el balance global de beneficio-riesgo se considera positivo, no será necesario llevar a cabo estudios de seguridad cardiovascular poscomercialización. Estas recomendaciones han tenido consecuencias inmediatas, y existen ya en marcha varios estudios de seguridad cardiovascular con nuevos AD45. De hecho, apenas 3 años después de que la FDA publicara sus recomendaciones, ya hay metaanálisis de seguridad cardiovascular publicados con nuevos AD46. La tranquilidad que estos han podido proporcionar no ha impedido, sin embargo, que en algún caso se demanden estudios adicionales, incluso en aparente ausencia de señal de alarma, con sorpresa no solo para el promotor sino para muchos expertos45. Y por supuesto, hay que recordar la necesidad, en absoluto fácil, de asegurar un seguimiento a largo plazo de los estudios suficientemente completo, ya que las pérdidas de seguimiento pueden dar al traste con la fiabilidad de un estudio43. En Europa, las recomendaciones –aún no finales- de la EMA14 van en la misma dirección de excluir, antes de la aprobación, cualquier exceso de riesgo cardiovascular asociado al AD, aunque son bastante menos concretas que las norteamericanas en cuanto a definir requerimientos específicos o límites para el exceso de riesgo. Se sugieren seguimientos suficientemente largos, inclusión de población de riesgo suficiente y necesidad de un análisis integrado de todo el programa de fase II y III –en la línea del solicitado por la FDA- antes de la aprobación que permita descartar riesgos excesivos. La EMA advierte que un incremento en ciertos eventos (no se especifica su cuantía) puede inducir, como en EE. UU., el requerimiento de un estudio específico de seguridad cardiovascular que excluya definitivamente el posible exceso de riesgo. En cuanto a las variables, la recomendación principal es, como en la FDA, un MACE compuesto por mortalidad cardiovascular, infarto de miocardio no fatal y ACV. La EMA advierte sin embargo que se evaluarán también otros eventos, como isquemia miocárdica (término quizá un tanto ambiguo por excesivamente amplio en cuanto a la diversa gravedad de lo que puede comprender, desde isquemia silente hasta angina inestable), hospitalización por síndrome coronario agudo, revascularización y/o empeoramiento de la insuficiencia cardiaca. Asimismo se recuerda la necesidad de recoger de forma sistemática efectos como incremento de peso, retención de líquidos y edema, cambios relevantes en la función cardiaca (ecografía, BNP/NT-pro BNP), tensión arterial o ritmo cardíaco. Cabe anotar, sin embargo, que la EMA contempla además la posibilidad, y ofrece recomendaciones generales al respecto, de que pueda investigarse el retraso en la aparición de diabetes para pacientes con intolerancia oral a la glucosa o con glucemia alterada en ayunas. Se valora también la posibilidad de estudiar el posible retraso en la aparición de complicaciones de la diabetes, tanto en lo referido al beneficio cardiovascular como a otras complicaciones, como retinopatía o nefropatía.
ConclusionesEl desarrollo clínico de nuevos AD se encuentra en un proceso de cambio radical aún por completar. Las agencias reguladoras, recogiendo la demanda del mundo científico y de los pacientes, y manteniendo sabiamente alejado el nada riguroso ambiente mediático, han emitido recomendaciones al respecto que parecen razonables y que, previsiblemente, redundarán en más y mejor información disponible para los nuevos AD en el momento de su comercialización, y en una posterior actualización periódica más completa. De esta forma, pacientes y médicos pueden tener en todo momento la garantía de que la seguridad de los nuevos AD está debidamente controlada y monitorizada. Es, sin embargo, evidente que estos nuevos requisitos complican, alargan y encarecen de forma considerable el desarrollo clínico de nuevos AD. Eso podría motivar que algunos abandonen el empeño de investigar en este campo, pero un vistazo al panorama actual sugiere que, en general, la cantidad y variedad de nuevos AD explorados es variado y amplio, con nuevas opciones como nuevos antiinflamatorios (inhibidores NF-kB o antagonistas de la IL-1) activadores de las sirtuinas y otros, para los que se valora la posibilidad no solo de no perjudicar, sino en lo posible reducir, la carga de complicaciones cardiovasculares asociada a la diabetes47. Cabe insistir, sin embargo, en la complejidad de este propósito y, como se ha señalado, “mantener la vista en la pelota”22. Incrementar la seguridad de los AD y la amplitud y longitud de los ensayos clínicos con nuevos AD redunda en beneficio de todos. Exigencias excesivas, poco o nada viables, podrían terminar con la investigación en ese campo, riesgo que por fortuna parece, al menos de momento, alejado. Queda además por ver, como señalé anteriormente, si estos nuevos requisitos satisfarán las expectativas de los responsables del reembolso y financiación de los medicamentos, algo que cada vez tiene más impacto en la forma y amplitud del desarrollo de nuevos fármacos en todos los campos48,49. En este sentido no sería extraño que el futuro pase por estudios comparativos de efectividad50, pero ese es ya otro cantar.
Conflicto de interesesEl autor es empleado de GlaxoSmithKline S.A. y posee acciones de dicha compañía. GlaxoSmithKline tiene medicamentos en investigación para el tratamiento de la diabetes tipo 2.
