





Alexander disease is a rare form of leukodystrophy that involves mainly astrocytes; it is inherited in an autosomal recessive manner and occurs by mutations in the GFAP gene, located on chromosome 17q21. It can occur at any age, and its infantile form is characterized by macrocephaly, seizures, severe motor and cognitive delay, and progressive spasticity or ataxia.
Case reportAn 8-month-old female was evaluated with a history of neurodevelopmental delay and unprovoked focal motor seizures. Physical examination showed normal head circumference, increased motor responses to tactile and noise stimuli, pyramidal signs and no visceromegaly. The widespread hypodense white matter was found on magnetic resonance, and lumbar puncture showed hyperproteinorrachia. Krabbe disease was ruled out by enzymatic assay and sequencing of GALC gene. In the reassessment of the case, abnormalities in neuroimaging lead to suspicion of Alexander disease, and GFAP gene sequencing reported a pathogenic mutation in exon 4 c.716G>A, which caused a change of arginine to histidine at position 239 of the protein (p.Arg239His).
ConclusionsThe neuroradiology signs observed in the resonance were decisive for the diagnosis later confirmed by molecular techniques. It is important to consider that certain mutations are not associated with macrocephaly, which may cause a delay in the diagnosis.
La enfermedad de Alexander consiste en una forma de leucodistrofia poco frecuente que afecta principalmente a los astrocitos; tiene un patrón de herencia autosómica recesiva y es causada por mutaciones en el gen GFAP, localizado en el cromosoma 17q21. Puede presentarse a cualquier edad y la forma infantil se caracteriza por macrocefalia, crisis convulsivas, retraso motor y cognitivo grave y espasticidad o ataxia progresivas.
Caso clínicoFemenina de 8 meses evaluada por retraso psicomotor y crisis convulsivas motoras focales no provocadas. En la exploración física, con perímetro cefálico normal, respuesta motora incrementada ante estímulos táctiles y al ruido, signos piramidales y ausencia de visceromegalias. Se observó hipodensidad generalizada de la sustancia blanca en la resonancia magnética y punción lumbar con hiperproteinorraquia. Se descartó enfermedad de Krabbe mediante ensayo enzimático y secuenciación del gen GALC. En la reevaluación del caso, las alteraciones en la neuroimagen hicieron sospechar de enfermedad de Alexander, y la secuenciación del gen GFAP reportó una mutación en el exón 4 c.716G>A, lo que ocasionó un cambio de arginina por histidina en la posición 239 de la proteína (p.Arg239His).
ConclusionesLos signos radiológicos en la resonancia fueron determinantes para el diagnóstico, que posteriormente se confirmó con estudio molecular. Es importante considerar que ciertas mutaciones no se asocian con macrocefalia, lo cual puede ocasionar retraso en el diagnóstico.
Alexander disease (OMIM #203450) is a neurodegenerative disorder part of the infantile leukodystrophy group. It is extremely rare and mainly affects the astrocytes in the hippocampus, striatum nucleus, and neocortex.1,2 Also, Alexander disease arises by mutations in the glial fibrillary acidic protein gene (GFAP), which is located on chromosome 17q21 and has an autosomal dominant pattern; it occurs most frequently in females.3,4
Considering the age at which the disease occurs, there are four varieties: neonatal, infantile, juvenile and adult.5 The neonatal form begins in the first year of life; it is rapidly progressive, with seizures, hydrocephalus, severe motor and cognitive retardation and progressive spasticity or ataxia. Patients die within the first two years of life.6 The infantile form is the most common and occurs in 51% of the cases, usually during the first two years of life, with similar symptoms and early death, although some patients survive until adulthood. One of the cardinal manifestations of this variant is macrocephaly related to megalencephaly.7,8 The juvenile form (23%) begins between four and ten years of age. The affected patients show bulbar or pseudobulbar signs like frequent vomiting, language and swallowing problems, gradual loss of cognitive functions and the rest of the other variants-like manifestations. Survival is up to 20 to 30 years of age.8,9 In the adult form (24%), the manifestations are highly variable, with bulbar or pseudobulbar signs, pyramidal signs, cerebellar dysfunction, dysautonomias, sleep disorders and seizures.10–12
This article describes a case with Alexander disease without macrocephaly. Furthermore, the main aspects of the differential neuroradiology and molecular diagnosis of leukodystrophies are revised.
2Clinical caseFemale patient of 8 months of age, with no consanguineous parents, a product of the second pregnancy, no perinatal history. She made visual contact at two months of age, social smiling at four months, failed to support the head. At seven months of age, she presented focal motor tonic-clonic seizures on the right side of the body, unprovoked origin. Treatment with magnesium valproate was initiated without improvement. Clonazepam was added, and because of the persistence of the crisis, the patient attended to our institution. During the physical examination, she showed normal head circumference (p25), appropriate eye contact, normal fundus, motor response increased to tactile and noise stimuli, hypertonia and generalized hyperreflexia, Babinski reflex and substitutes present and without visceromegaly.
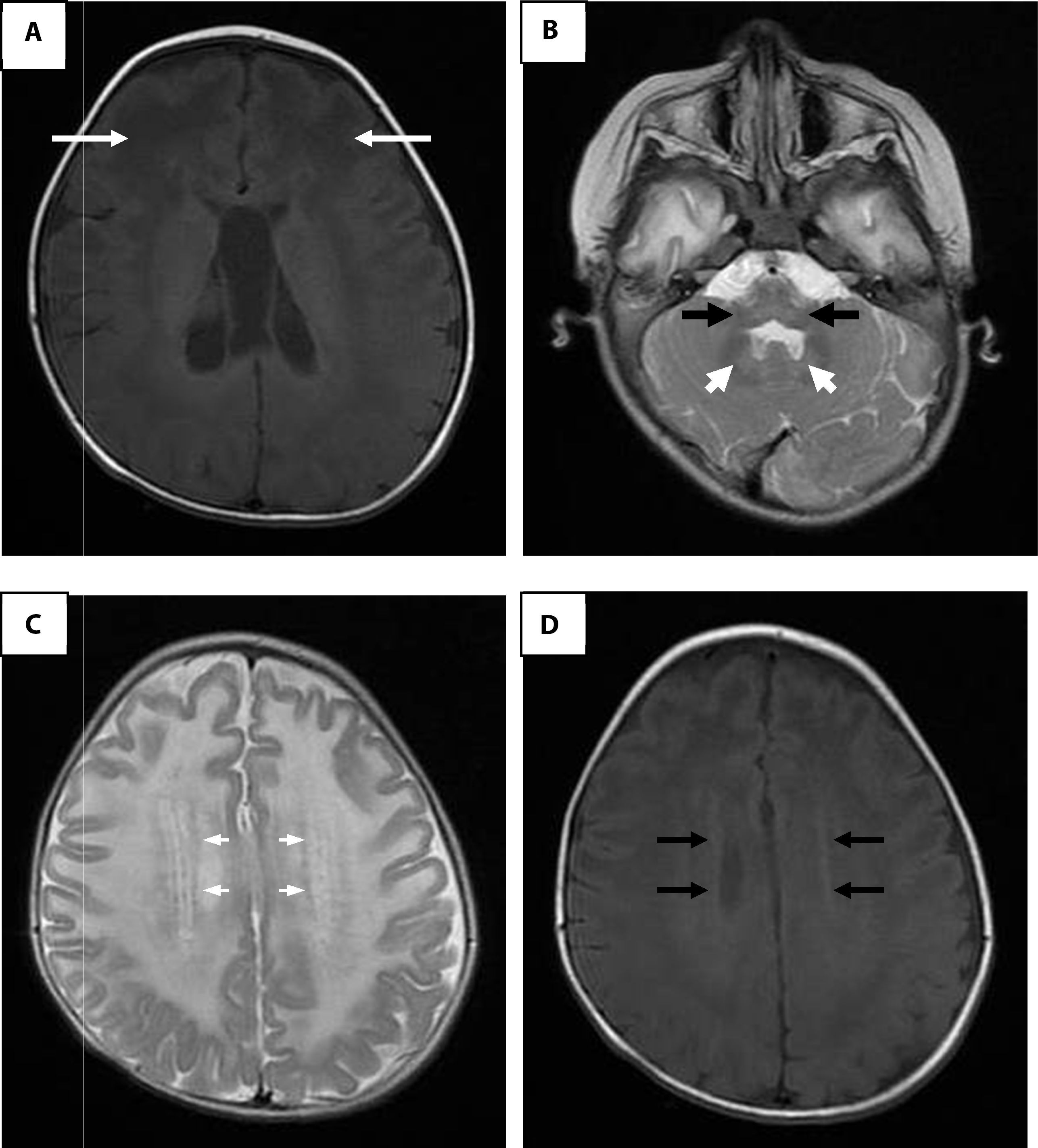
Lumbar puncture showed hyperproteinorraquia. Computed cranial tomography showed widespread white matter hypodensity. The magnetic resonance imaging (MRI) of the brain corroborated the condition of generalized white substance, thalami, brain stem, basal ganglia and cerebellum (Fig. 1). Krabbe disease was discarded by enzyme assay and GALC gene sequencing. The case was re-evaluated when the patient was 20 months of age. The alterations in the MRI made suspicious of Alexander disease, and the sequencing of the GFAP gene reported a mutation in exon 4 (c.716G>A), which resulted in a change of arginine by histidine at position 239 of the protein (p.Arg239His).
 in T1 where arrows indicate extensive white matter changes of frontal predominance. B. White matter signal alterations of the cerebellum, dentate nucleus and brainstem in a fast SE (FSE) T2 enhanced image. C. Hyperintense rim in the axial projection in FSE T2. D. Hypointense rim in the axial projection in T1.")
Brain magnetic resonance imaging. A. Enhanced image in axial projection spin-echo (SE) in T1 where arrows indicate extensive white matter changes of frontal predominance. B. White matter signal alterations of the cerebellum, dentate nucleus and brainstem in a fast SE (FSE) T2 enhanced image. C. Hyperintense rim in the axial projection in FSE T2. D. Hypointense rim in the axial projection in T1.
Leukodystrophies are a group of inherited diseases that affect the white matter of the central nervous system and occasionally the peripheral nervous system. They cover about 40 entities, and their differential diagnosis in children is complex since it requires a systematic analysis that begins with the completion of a proper medical history and a complete neurological examination. The family history and the family tree can guide towards recessive disorders linked to the X chromosome, where affected maternal uncles or autosomal recessive forms in families with consanguinity or inbreeding exist.13,14
In the majority of leukodystrophy cases, there is a period of static or progressive normal development (excluding early neonatal or infantile forms) before the loss of skills. The onset of the symptoms in Alexander disease may occur at any stage of life, unlike other entities such as Krabbe disease, which develops in early childhood.8,13
Leukodystrophies usually are not dysmorphic, except in some cases. For example, in Canavan disease and Alexander disease macrocephaly is observed, and it is even considered as a distinctive hallmark of these disorders. In the deficiency of the transporter of thyroid hormones MCT8, symptoms as movement disorders like dystonia or dyskinesia are characteristic. In Krabbe disease, peripheral neuropathy is very common. Adrenoleukodystrophy presents metachromatic leukodystrophy, low academic achievement, and attention disorders. In both the leukoencephalopathy with vanishing white matter and the 4H (hypomyelination, hypogonadotropic, hypogonadism and hypodontia) syndromes, ataxia predominates. Other leukodystrophies develop late seizures; however, this may be the earliest manifestation of Alexander disease.13,14 The present case showed a variety of infantile onset, with developmental delay, severe hypotonia, and seizures without macrocephaly appearance.
The analysis and search for signs in the MRI are determining factors for the differential diagnosis of diseases that affect the white matter. The relevancy in the imaging study is to establish if an abnormality is a demyelination or hypomyelination depending on the intensity of the signal, the identification of the region, to determine if it affects specific tracts and if the patient has confluent or multiple injuries. If the pattern corresponds to a hypomyelination and a slowly progressive entity, the Allan-Hernon-Dudley syndrome due to the deficiency of thyroid hormone transporter MCT8 should be considered. The absence of cortical atrophy is characteristic of Pelizaeus-Merzbacher disease, whereas the cerebellar atrophy corresponds to the 4H syndrome. Diffuse white matter involvement is typical of megalencephalic leukoencephalopathy with subcortical cysts.
The affected regions may lead to the diagnosis: the parietooccipital region, to adrenoleukodystrophy or Krabbe disease; the temporal region, to Aicardi-Goutières syndrome; the subcortical region, to Kearns-Sayre syndrome; the periventricular region, to metachromatic leukodystrophy, and the frontal region, to Alexander disease. In Krabbe disease, symmetrical hyperintensities in the optic nerve and the pyramidal tracts are observed.13–15
It has been established that the identification of four of the five characteristic radiological findings of Alexander disease can give a greater certainty in the diagnosis (Table 1).16 In the present case, four criteria were found; the fifth was not evaluated because the study was not available. Adult and juvenile forms of Alexander disease may not have very distinctive findings, such as larger lesions of posterior fossa structures, stem atrophy and diffuse signal changes of basal ganglia and thalami. Some authors propose that the alterations of posterior fossa structures, atrophy and tumor-like lesions in the brain stem, abnormalities in the thalamus, nuclei of the base, bulb or spinal cord signals, cobblestone organization of ependymal cells and gadolinium uptake by the injuries are sufficient to suspect of an atypical form of Alexander disease.17,18
Alexander disease magnetic resonance imaging criteria.
| 1) Extensive white matter changes with frontal predominance |
| 2) Periventricular hypodense rim in T2 and hyperintense in T1 |
| 3) Abnormalities of basal ganglia and thalami, which include edema, signal hyperintensity or hypointensity at T2 sequence |
| 4) Anomalies in the rhombencephalon (hindbrain), particularly in the brainstem and the mesencephalon (midbrain). |
| 5) Reinforcement with gadolinium of the periventricular rim, frontal white matter, fornix, optic chiasm, base nuclei, thalami, dentate nucleus, and brainstem nuclei |
Before the molecular diagnosis, the presence of protein deposits (known as Rosenthal fibers) in a brain biopsy was the only way to confirm Alexander disease. During the disease, these fibers composed of glial fibrillary acidic protein (GFAP) aggregates, vimentin, αβ-crystallin and heat shock protein-27 increase in size and number. GFAP is a 432 amino acids cytoskeleton component that provides structural stability to the mature astrocyte.19–21
More than 100 mutations in the GFAP gene that affect the protein with a dominant negative effect have been described. The mutant protein prevents the function of the wild protein through a toxic mechanism that is not yet fully understood.2 It is postulated that there is not a direct effect on the protein synthesis, but that the mutant protein affects the oligomerization process or the solubility. Abnormal oligomerization may also inhibit the proteasome activity in the astrocyte in such a way that it affects the normal interaction between astrocytes and oligodendrocytes, resulting in hypermyelination or demyelination. Immunohistochemistry has demonstrated a decrease in the glutamate transporter-1 levels in hippocampal astrocytes. In addition, different studies have shown that the decrease in glutamate capture has a very important role in neuronal damage. The extracellular glutamate excess increases the excitatory neuronal activity, and it manifests as very difficult-to-control seizures.22
The majority of the genetic variants of Alexander disease correspond to missense mutations (especially Arg79, Arg88, and Arg239), and are found together in 34% of the cases. The pathogenic more recurrent variants exclusive of the infantile, juvenile and adult forms are presented in Table 2. Some mutations have been described in all the forms of the disease, such as p.Arg416Trp.8
Pathological mutations specific to each form of Alexander disease.
| Infantile form | Juvenile form | Adult form |
|---|---|---|
| p.Met73Thr | p.Leu235Pro | p.Arg66Gln |
| p.Leu76Phe | p.Arg70Trp | |
| p.Asn77Ser | p.Arg70Gln | |
| p.Lys86_Val87delinsGluPhe | p.Met74Thr | |
| p.Leu97Pro | p.Glu205Lys | |
| p.Arg239His | p.Leu359Pro | |
| p.Arg239Leu | p.Ala364Thr | |
| p.Leu352Pro | ||
| p.Glu373Lys | ||
| p.Asp417MetfsTer15 |
In the present case, a mutation in exon 4 (c.716G>A) that affects the coiled-coil protein domain 2A was demonstrated. Most common mutations are found in exon 1 (45%), followed by exon 4 (27.2%) and 6 (16%). According to Prust et al., no mutations were found in exons 2 and 9 in a study of 215 cases.5 The authors proposed the existence of two phenotypes based on the age of onset: early onset type 1 (or cerebral), and late onset type 2 (or bulbar). In addition to classic neuroradiology findings described by van der Knaap et al.16, type 1 includes the most typical cases with macrocephaly, growth arrest, encephalopathy, seizures, cognitive and motor delays in the first four years of life. Type 2 displays an unusual case where bulbar signs, autonomic dysfunction, cerebellar signs, dysphonia, abnormalities in eye movements and palatal myoclonus prevail. In this phenotype, abnormalities in the white matter of the posterior fossa, brain stem, cerebellum and spinal cord atrophy are typical.
In the present case, the arginine residue is located at position 239, which turns out to be a frequent site of mutation (20.3% of cases), as well as being highly conserved between species. Clinical cases that carry this mutation have the cerebral phenotype of the disease, and most of them show macrocephaly. In Alexander disease, this disorder often develops quickly, perhaps as a result of the proliferation of atypical astrocytes. It is possible that cases without macrocephaly have a disruption of the cell energy balance that prevents neuronal proliferation. The comparison of cases with Arg239 condition showing early onset age and psychomotor retardation is shown in Table 3.5 It is clear that the case reported showed no macrocephaly or neuro-regression.
Comparison of cases with the Arg239 condition.
| Goyal et al.23 | Caroli et al.24 | Present case | |
|---|---|---|---|
| Differential age | 16 months | 4 months | 8 months |
| Seizures | + (4 months) | + (NR) | + (7 months) |
| Psychomotor retardation | + | NR | + |
| Regression | + | NR | - |
| Spasticity | + | + | + |
| Hyperreflexia | + | + | + |
| Macrocephaly | - | + | - |
| MRI findingsa | 1,2,3 | 1,2,3,4 | 1,2,3,4 |
NR, not reported; MRI, magnetic resonance imaging.
MRI findings obtained from van der Knaap et al.: (1) extensive white matter changes, (2) periventricular rim, (3) abnormalities in basal ganglia and thalami, (4) abnormalities in brainstem, and (5) reinforcement with gadolinium [Modified from Ref. 16].
Alexander disease is a disorder that shows classic, distinctive manifestations. However, it is important to consider it as a differential diagnosis in atypical cases without macrocephaly or neuro-regression. MRI data may be useful in the differential diagnosis against other leukodystrophies, especially with an early onset of symptoms, even when macrocephaly is not present.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
FundingNone.
Conflict of interestThe authors declare no conflicts of interest of any nature.
To Dr. Merjo van der Knaap for her valuable contributions to the clinical and radiological analysis of the patient.
Please cite this article as: Esmer C, Villegas-Aguilera M, Morales-Ibarra JJ, Bravo-Oro A. Presentación atípica de la enfermedad de Alexander infantil sin macrocefalia. Bol Med Hosp Infant Mex 2016;73:196–201.