


Malignant peripheral nerve sheath tumors (MPNSTs) are aggressive sarcomas that occur in adulthood and are located mainly on the trunk and lower limbs, with a high association with neurofibromatosis type 1 (NF1).
Case reportA 34-months-old female infant without NF1 with a palpable abdominal tumor is described. The tumor corresponded to a retroperitoneal MPNST. The diagnostic approach and management are presented, highlighting the complications and sequelae during the evolution of the patient.
ConclusionsDespite their low incidence, MPNSTs are important because of their aggressiveness, and should be considered upon the detection of a tumor located at paravertebral level or limbs, especially in patients with NF1. The cornerstone of the treatment lies in a complete surgical resection due to the high rate of recurrence and limited therapeutic response to radiotherapy and chemotherapy. This case presents clinical manifestations and complications that can be expected with these tumors and their harmful behavior. The absence of NF1 does not exclude the diagnosis.
Los tumores malignos de la vaina de nervio periférico (MPNST, por sus siglas en inglés) son sarcomas raros y agresivos que aparecen principalmente en la edad adulta; se localizan principalmente en tronco y extremidades inferiores, con una alta asociación con neurofibromatosis tipo 1 (NF1).
Caso clínicoSe describe el caso de una niña de 34 meses de edad sin NF1, quien consulta por masa abdominal. La masa correspondió a un MPNST retroperitoneal. Se presenta el abordaje diagnóstico y la conducta terapéutica, resaltando las complicaciones y las secuelas que se presentaron.
ConclusionesLos MPNST, a pesar de su baja incidencia, son importantes debido a su agresividad y deben sospecharse ante una masa localizada a nivel paravertebral o en extremidades, en especial en pacientes con NF1. La piedra angular en el tratamiento es la resección quirúrgica completa, debido a la alta tasa de recidiva, y una respuesta terapéutica a la radioterapia y quimioterapia limitada. Este caso muestra las manifestaciones clínicas y las complicaciones que se pueden esperar con estos tumores, así como su comportamiento agresivo. La ausencia de NF1 no descarta el diagnóstico.
Malignant peripheral nerve sheath tumors (MPNSTs) are sarcomas derived from nerve sheath Schwann cells or pluripotent neural crest cells.1 They are also known as neurilemmoma, malignant schwannomas, neurofibrosarcomas or neurogenic sarcomas.2 MPNST are very rare tumors with an incidence of 0.001% in the general population and 0.16% of patients with neurofibromatosis type 1 (NF1).3 They represent approximately 10% of soft tissue sarcomas. Despite their rarity, they are one of the most frequent non-rhabdomyosarcoma tumors diagnosed in pediatrics.4 They are highly aggressive and associated with NF1 and previous radiation therapy.5
The aim of this paper was to present a case of a retroperitoneal MPNST in a patient without neurofibromatosis. Also, a review of the literature was conducted, and the complementary therapeutic options after surgical treatment are discussed further.
2Clinical caseA female patient of 2 years and ten months of age who lived in an urban residence arrived with an abdominal mass identified by her mother seven months earlier, which had an accelerated growth during the last two months. No pain, fever, weight loss or any other associated symptoms were present. She had a history of long-standing chronic constipation. There was no family history of neurofibromatosis or cancer. Physical examination revealed a 15 x 10cm fixed painless mass of hard consistency in the left lower abdominal quadrant. No café-au-lait macules were present, neither any other signs that suggested neurofibromas. Blood count, lactate dehydrogenase, uric acid, serum electrolytes, and renal and liver function tests were normal. Alpha-fetoprotein and beta subunit of human chorionic gonadotropin were normal, discarding any germ cell tumor.
Abdominal ultrasound reported a large, macrolobulated retroperitoneal tumor of 90 x 64 x 78mm in its lateral, anteroposterior and transverse diameters, with heterogeneous echogenicity but predominantly hypoechoic. No enlarged retroperitoneal lymph nodes were identified.
Abdominal computed tomography (CT) showed a heterogeneous tumor in the left iliac fossa, adjacent to the ipsilateral L5-S1 intervertebral foramen. The chest X-ray was normal.
Laparotomy was performed, and an incisional biopsy was taken. Gross appearance of the tumor was suggestive of a fibrosarcoma, but the sample was insufficient for diagnosis; thus, a new biopsy was taken. A hard, well delimited retroperitoneal 15 x 10cm mass, with a large vascular component which emerged from the back adjacent to the lumbar vertebrae and iliac vessels was described. It compressed the pelvic organs forward. The abdominal cavity was free of tumor and contained clear fluid.
Histopathological analysis revealed an atypical spindle cell neoplasia of difficult classification, with no evidence of necrosis and with a mitotic count up to three per 10 high-power fields. It was not possible to define its histopathological origin with certainty.
Immunohistochemistry was carried out in the pathology department of the Hospital Universitario Fundación Santa Fe de Bogotá (FSFB). Tumor cells showed high diffuse reactivity for tumor protein markers S100 and CD34, and occasional focal positivity for PGP 9.5 and synaptophysin. The rate of cell proliferation (Ki67) was 5%. Cells were negative for GFAP, HMB45, EMA, CK7, AE1/AE3, TLE1, ALK, desmin, CD117, CD99, and DOG1.
Samples were also analyzed at Brigham and Women's Hospital (BWH) Department of Pathology, Boston, USA. A fusiform cell neoplasm, of moderate cellularity, in which tumor cells had a pale eosinophilic cytoplasm and slightly ovoid or fusiform nuclei was reported. Multifocal scattered mitosis. No significant pleomorphism or necrosis was present. Immunostains showed multifocal positivity for S-100 protein, weak focal positivity for SMA and multifocally scattered MDM2 positive cells, negative for GFAP. Morphological and immunophenotypic findings suggested a malignant tumor of the peripheral nerve sheath, which appeared to be of low-grade.
The patient did not meet the clinical criteria or a family history of NF1; ophthalmological examination was normal, and the clinical geneticist did not consider necessary to do specialized genetic studies.
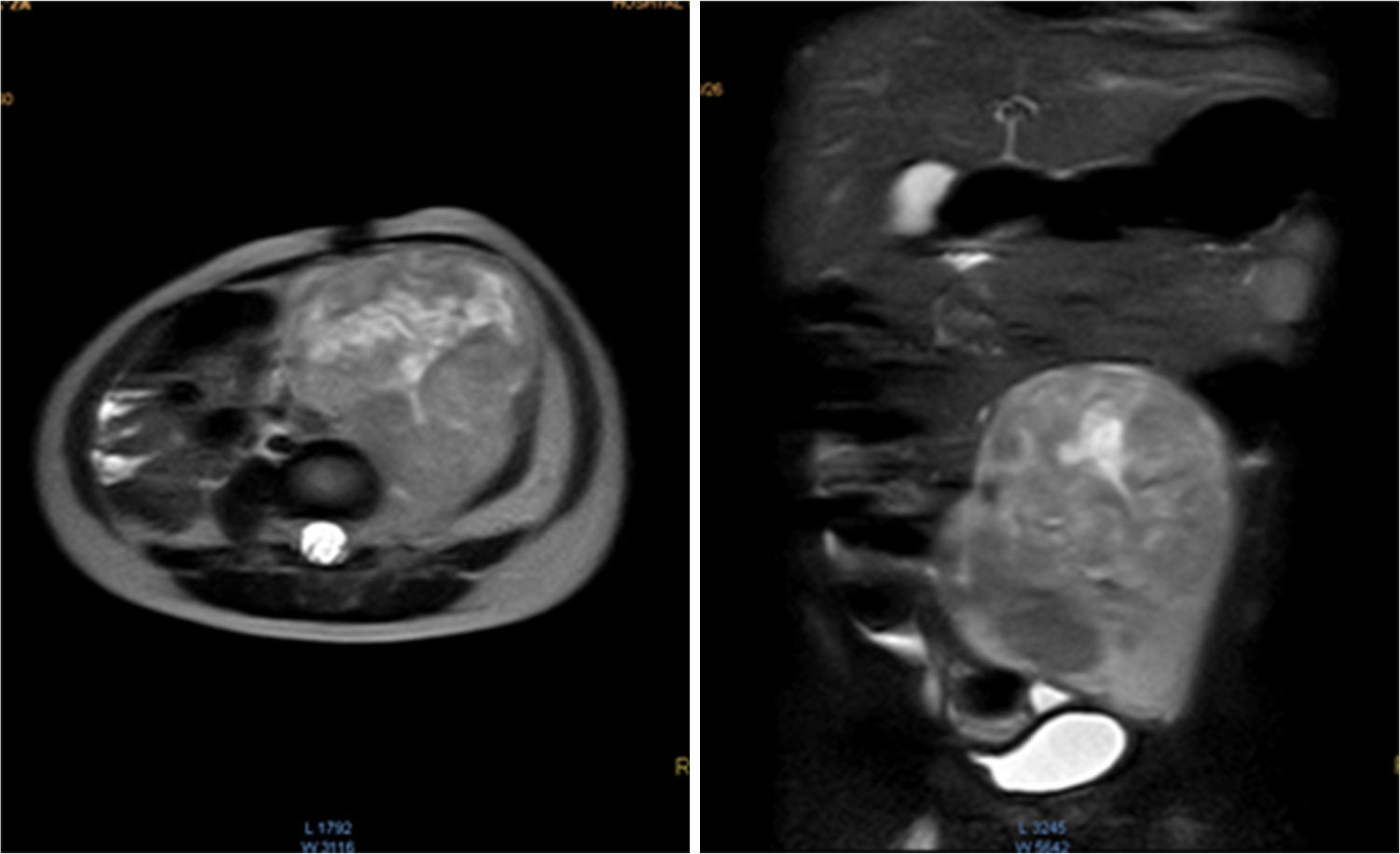
Magnetic resonance (NMR) was performed before tumor resection. The results showed that the tumor surrounded the left iliac artery and vein (Fig. 1).

Contrasted abdominal MRI is showing a solid mass of 107 x 80 x 76mm in its craniocaudal, anteroposterior and transverse diameters, which occupies hypogastrium, left iliac fossa, left flank and mesogastrium. It is pedunculated and comes from left L4-L5 and L5-S1 intervertebral foramina. It is solid, heterogeneous, vascularized, with necrosis. It partially surrounds the aortic bifurcation and totally the left iliac artery and its branches, so as the iliac vein.
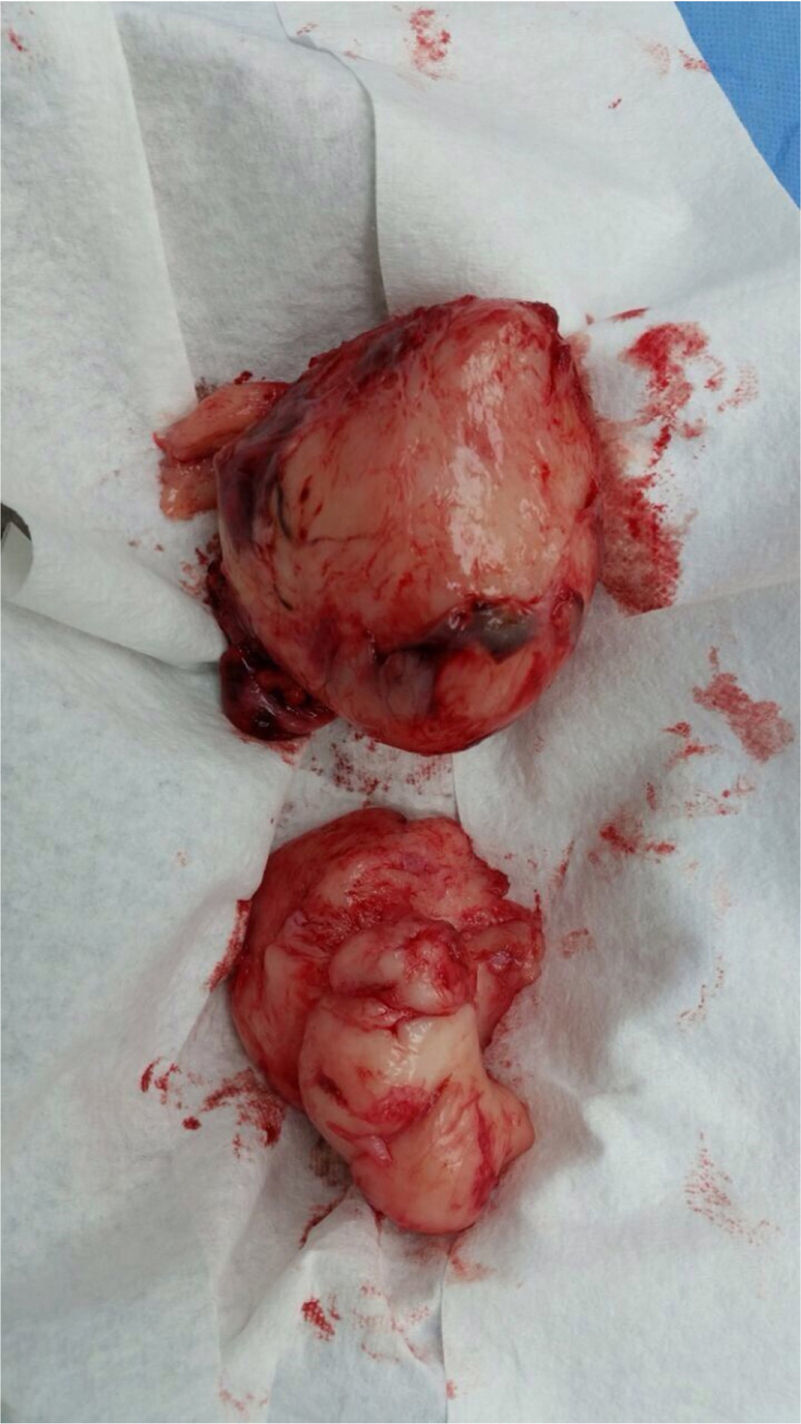
Surgery was performed again to excise the tumor. The tumor surrounded the left iliac artery, ureter, gonadal artery and femoral vein. About 80% of the tumor was resected (Fig. 2). During surgery, the patient presented a hypovolemic hemorrhagic shock. Therefore, red blood cell transfusions and vasopressors were necessary. The patient was admitted to the Intensive Care Unit, where she stayed for five days. The histopathologic report was similar to the one described above but with a proliferation index of 15% measured with Ki67.

Postoperative pain and edema in the left thigh, muscle weakness, limitation to adduction, dysesthesia of the back of the foot and clubfoot which impaired gait were present; vascular compromise was ruled out by Doppler ultrasound. S1 nerve lesion in addition to physical unfitness was found. The patient began with physical therapy and splinting. Electromyography reported L5-S1 root lesion vs. sciatic nerve injury. Currently, the patient has recovered significantly. She has a slight limp, foot drop, hypotrophy, and paresis of the affected limb. She continues with physical therapy.
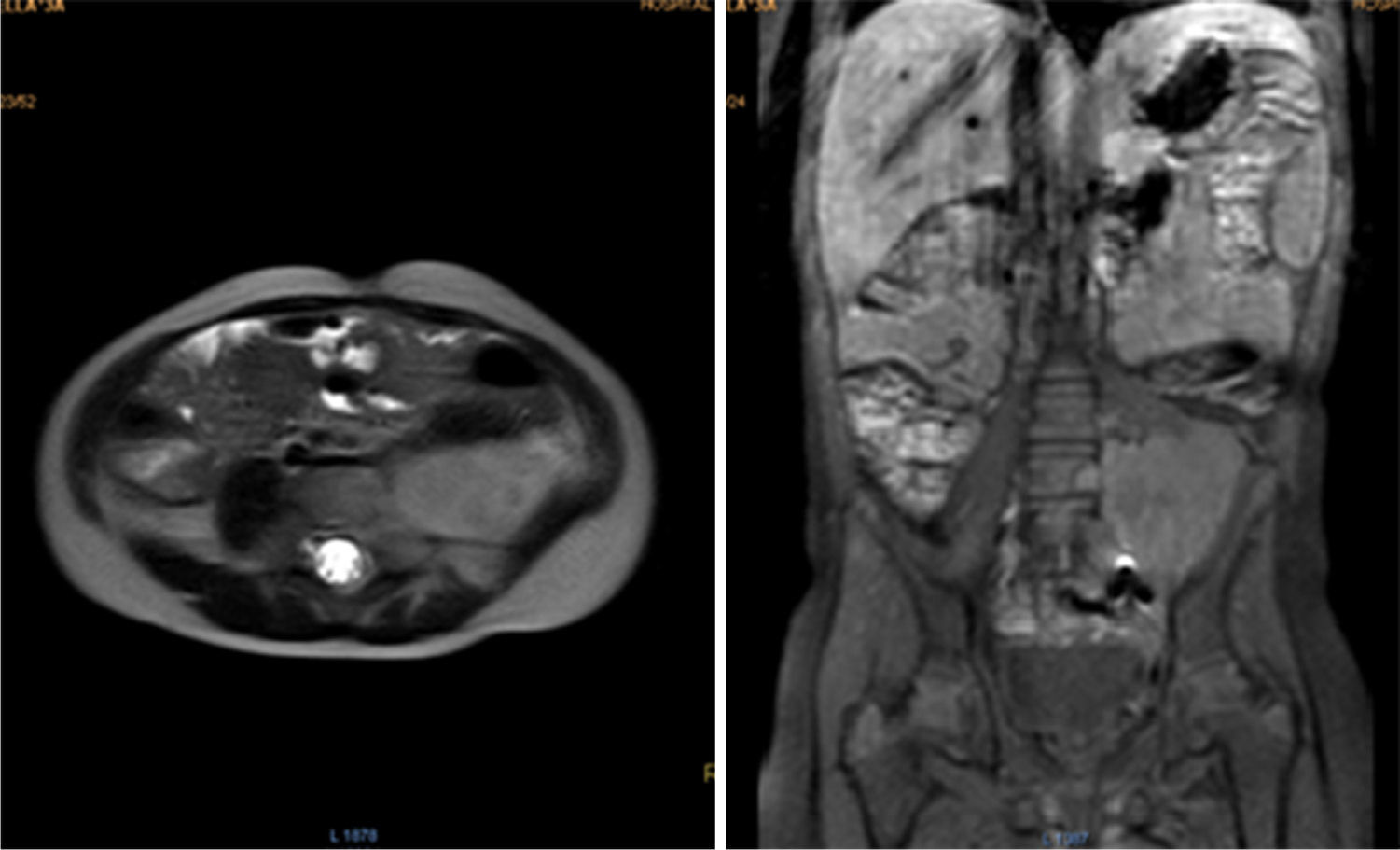
During oncologic follow-up, laboratory tests have been normal. Postoperative abdominal MR reported a 42.2 x 53.6mm residual mass (Fig. 3). Three months later, ultrasound showed an apparent increase in tumor size (73 x 54 x 58mm). Chemotherapy consisted in ifosfamide plus doxorubicin in alternation with ifosfamide plus etoposide.

Further therapy will be decided according to her evolution. The parents consent to the publication of this case, keeping the confidentiality of the patient.
3DiscussionMPNST are rare tumors that occur more frequently in 20- to 50-year-old patients. The cases reported in children are very scarce.4–6 The patient in this report presented a palpable abdominal tumor since she was 27 months of age, which was diagnosed at 34 months, age at which this disease is extremely rare. Bates et al.7 found a total of 139 reported cases of MPNST in patients under 19 years of age between 1973 and 2009, and only 5 cases in children between 1 and 4 years of age (3.6%) in the SEER (Survival Epidemiology and End Results) database. This age group had a lower incidence (0.19 per million person-years, 95% CI 0.09 to 0.36), with a statistically significant difference with the 10 to 19-year-old group (10- to 14-year-old group with an incidence of 0.72, 95% CI: 0.52 -0.97; and the 15- to 19-year-old group with an incidence of 0.99, 95% CI: 0.75-1.28, p < 0.001). In the study, no significant differences between ethnic groups were found, although no specific data on Hispanics were mentioned.
Another study, which also used the SEER database, compared children and adults with MPNST from 1973 to 2008.1 This study reported that the percentage of non-Hispanic white adults was 68.3%, whereas there was a greater proportion of African-American and Hispanics compared to non-Hispanic whites in children (18.8%, 26.6%, and 49.2%, respectively) with a statistically significant difference (p < 0.0001).
From the clinical point of view, literature reports include slow-growing tumors, prolonged asymptomatic periods, and late onset start of signs of the disease and sensorimotor disturbances.9 In this patient, the disease presented as an asymptomatic mass of slow growth without neurological findings.
The most accurate image studies are CT and MRI, over ultrasonography. CT is useful in assessing the extent of the tumor and metastases, but MRI is the test of choice because it can reveal the nerve of origin and its relationship to vascular structures, muscle, and surgical landmarks.8,10,11 The CT scan performed on the patient showed a close relationship with the L5-S1 intervertebral foramen, which was subsequently confirmed with MRI and consistent with the knowledge that most MPNST originate from major nerve trunks, including the sciatic nerve, brachial plexus, and sacral nerve. Hence, the most common locations are the trunk and proximal areas of the extremities.12,13 A useful image is positron emission tomography (PET scan), which displays the metabolism of glucose by tumor cells and is primarily helpful for demonstrating the malignant transformation of plexiform neurofibromas.9
The tumor (100 x 150mm) was larger than the median size reported in children with chest or abdomen MPNST in the United States, and was close to the larger sizes reported in that country (median 85mm with a standard deviation of 67mm). It was found that mortality was higher in females, probably due to larger tumor sizes (median 85mm) compared with males (median 63mm).1 Females (57%) had tumors of at least 70mm, which only occurred in 26% of males; the cause of this difference is unknown. In the reported patient, the mass location around the iliac artery, ureter, gonadal artery and left femoral vein made total resection impossible. Follow-up ultrasound suggested a significant tumor growth (73mm). Larger MPNST may be difficult to resect completely. These difficulties are reflected by the increased mortality of pediatric patients with stage III/IV tumors of the trunk (HR: 12.53, 95% CI 3.11-50.47) or of those classified as non-localized tumors because of regional spread or distant disease (HR: 0.53, p <0.001).1 Case reports from various countries and regions may help to determine whether these tumors are larger in females compared with males.
From the macroscopic point of view, the MPNST are bulky tumors (> 10cm) of deep location. In a transverse section, they resemble the appearance of fish meat with a necrosis and hemorrhage foci. Microscopically, there are highly cellular lesions arranged in fascicles which may be confused with fibrosarcomas or hypercellular schwannomas, alternating with hypocellular myxoid areas that may seem benign.14 Cells may be fusiform (similar to Schwann cells) or round (morphologically resembling fibrosarcomas or round cell sarcomas), with scarcely prominent cytoplasm, hyperchromic nuclei with well-defined nucleoli,9,14 frequent mitoses and focal necrosis.15 Pathology reports from BWH and FSFB described fusiform cells with pale eosinophilic cytoplasm and atypical ovoid nuclei and scattered multifocal mitosis.
S-100 protein has been identified in about 50-90% of MPNST by immunohistochemistry, usually with a focal pattern.2,6,10,14–16 Therefore, it is the most often used marker to document the differentiation of peripheral nerve sheath, but it is also present in synovial sarcomas, fusiform melanomas, and cellular schwannomas. CD34 is present in some MPNST. CD56 and its protein gene product 9.5 (PGP 9.5) are considered sensitive antigens for MPNST.2 In this case, there was a multifocal staining for S-100, and focal positivity for synaptophysin and PGP 9.5. EMA and CK7 were negative; these two proteins together have high specificity for synovial sarcoma.2 AE1/AE3, CD99 and TLE1, frequent in this type of sarcoma, were also negative. The tumor showed weak focal positivity for SMA unlike leiomyosarcomas, which stain strongly for this antigen. Desmin, which is present in many leiomyosarcomas, was negative in this case.16
In a study of risk factors, Chinese researchers found that when the S-100 protein was negative or weakly positive, patients had higher recurrence rate and worse survival, with a statistically significant difference when compared with S-100 positive patients.17
All the reported studies about treatment agree that the main goal is the complete surgical removal of the tumor, with negative surgical margins, which leads to a better disease-free survival (DFS), lower recurrence rate and increased overall survival (OS).4,12–14,18–20 Although there has been much controversy regarding the use of chemotherapy for the treatment of MPNST, studies confirm its benefit, especially with the combination of doxorubicin and ifosfamide in different regimens that significantly improves DFS and OS in patients with high-risk tumors.21,22 Since the limitations of these studies are inherent to their retrospective design, it is suggested to conduct prospective randomized studies regarding the use of chemotherapeutic agents for greater clarity. The Italian-German group has confirmed the benefit of chemotherapy, especially in protocols which use ifosfamide. They also recommend neoadjuvant chemotherapy for patients with tumors that are impossible to be completely removed at the time of diagnosis, to reduce the size of the tumor and allow surgery with continued chemotherapy and radiotherapy.4
Similarly, the use of radiation therapy as an adjuvant treatment is controversial; however, different studies show greater local control with the use of radiotherapy, especially when there is microscopic disease, but their benefit when there is a macroscopic disease is uncertain.4,13,23 Radiation therapy could not be classified as a risk factor in patients with MPNST nor showed statistically significant improvement in survival.4,23 Surprisingly, in another study, patients who received radiotherapy with less than 45Gy had a higher frequency of recurrence than those who did not receive radiation.18 As with chemotherapy, prospective randomized studies are required to conclude on the benefit of this treatment.
Given the suboptimal results on OS and DFE with conventional therapies, and the chromosomal alterations found in many of the MPNST, researchers have directed their effort toward the use of target substances that can act eliminating tumor cells with fewer side effects. It is known that the gene responsible for NF1 is located on chromosome 1724 (17q11.225), and encodes for neurofibromin, which functions as a tumor suppressor gene strongly expressed in neural tissue, kidney, spleen, and bone.24 It also interacts with the Ras gene, maintaining its inactive conformation.25 NF1 mutations that generate a loss in neurofibromin expression are considered tumor promoter events since Ras, an oncogene responsible for proliferation, invasion, and metastasis of tumor cells is activated.25 Most transduction pathways of intracellular signals that lead to activation of Ras and other tumor promoting proteins are started by tyrosine kinase receptors located on the cell membrane. Therefore, inhibition of these receptors by available antagonist drugs (such as imatinib, dasatinib, sunitinib, sorafenib) may be of importance in the treatment,25 although preliminary results are not very encouraging. However, it is possible to identify candidates for molecular-directed therapy. Alaggio et al.26 studied the BIRC5/survivin gene in the 17q chromosomal region of tumor cells in patients with MPNST and found that their high values correlated significantly with tumor size and the likelihood of survival, which supports the concept that survivin can be considered as a prognostic marker and a target for therapeutic interventions. Similarly, Patel et al., who worked on MPNST tumor cells of mice and humans, identified the overexpression and amplification of the AURKA (Aurora Kinase A) gene, which objective is Ras, and demonstrated that the blocking of this gene decreased tumor growth in vitro and in vivo.27
Finally, histone deacetylases (HDAC) are a family of enzymes involved in gene expression, DNA repair and response to stress; all these processes are altered in neoplastic cells. Therefore, HDAC inhibitors have high antitumor activity.28 Inhibiting the HDAC8 isoform induces growth arrest in the S phase of the cellular cycle.29 The use of drugs which inhibit this enzyme in combination with chemotherapeutic agents—such as some antimetabolites (5-fluorouracil, gemcitabine, and cytarabine) that mainly act on the S phase—are very promising procedures to improve tumor control of MPNST.28,29 Different studies confirm that HDCAs are excellent targets for cancer treatment. HDAC inhibitors have demonstrated to be effective in a broad range of solid tumors and hematologic diseases, markedly decreasing the chemotherapy-related toxicity.30
MPNST are highly malignant, with a recurrence rate of 40-65% and a metastasis rate of 40-68%, which depend on the degree of histological malignancy; thus, they have an unfavorable prognosis.3 The most frequent sites of metastasis are the lung, bone, and pleura; therefore, a chest radiograph is useful as shown in our patient. However, CT scan and bone scintigraphy are the preferred studies for the evaluation of metastatic disease.6
About 25-50% of MPNST occur in patients with NF1, and 10-20% occur after therapeutic or occupational irradiation.31 The patient reported here was a sporadic case who did not meet the clinical criteria and neither had a family history of NF1, nor she received radiotherapy. Anghileri et al. reported that the main prognostic factors for survival were a recurrence of the disease, tumor size and location (trunk or limbs).32 Additionally, patients who have had positive surgical margins had 2.4 times higher risk of local recurrence. Moreover, half of the patients who received radiotherapy had more frequent local recurrences, although there was no statistical difference with those who did not receive radiotherapy.32 Interestingly, the presence of NF-1 did not affect survival as an independent factor, which was confirmed in a meta-analysis published by Kolberg et al.33 However, patients with NF-1 had larger tumors and an earlier onset.32 The patient presented in this report had two significant risk factors: tumor size and the presence of a postoperative residual tumor. Regardless of clinical prognostic factors, Norwegian researchers demonstrated alterations in chromosomes 10, 16 and X of neoplastic cells from MPNST patients. They found that gains in 16p or losses in 10q or Xq identified a very high-risk group with a 10-year survival of only 11% with a highly significant statistical difference (p <0.00005).34
Despite multimodal therapy, overall 5-year survival ranges from 35 to 50%.22,35 A survival of 43-59% is reported in the Italian-German pediatric series.4
Despite its low incidence, MPNST are important because of their aggressiveness and should be considered in any case with a paravertebral or extremity mass, especially in patients with NF1. The cornerstone of the treatment is the complete surgical removal due to its high recurrence rate, and its limited response to radiation and chemotherapy. This case shows the clinical manifestations and complications that can be expected with these tumors and their aggressive behavior. The absence of NF1 does not exclude the diagnosis.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
FundingAuthor's own-funds.
Conflict of interestThe authors declare no conflicts of interest of any nature.
To the Hospital Universitario de Santander.
Please cite this article as: Rueda Arenas E, Pinilla Orejarena A, García Corzo J, Lozano Ortiz D. Tumor maligno de la vaina del nervio periférico retroperitoneal en un niño preescolar. Bol Med Hosp Infant Mex. 2016;73:188–195.