


Background: Patients with Silver-Russell syndrome suffer from severe intrauterine and postnatal growth retardation, relative macrocephaly and body asymmetry, among other characteristics. It is caused by several genetic and epigenetic mechanisms in 11p15.5 in 40% of the cases and maternal uniparental disomy of chromosome 7 in 10%.
Methods: Twenty patients with a diagnosis of Silver-Russell syndrome who were seen at the HIMFG from 1998 to 2012, were evaluated according to international clinical criteria confirming the diagnosis in nine of the subjects.
Results: All patients showed intrauterine and postnatal growth retardation and short stature, both considered as major criteria of Silver-Russell syndrome. Relative macrocephaly was present in 78% of the patients and asymmetry in 33%. Other characteristics such as renal tubular acidosis were present > 50% of the cases.
Conclusions: The clinical diagnosis of Silver-Russell syndrome is complex. Short stature is the main reason for seeking medical attention and is helpful in the identification of a differential diagnosis. This situation underlines the importance of growth and development evaluation of all patients and particularly in those with short stature to identify those cases that may require molecular studies, with implications in management, prognosis and genetic counseling.
Introducción: El síndrome de Silver-Russell presenta restricción del crecimiento intrauterino y posnatal, macrocefalia relativa y asimetría, entre otras características. Es causado por mecanismos genéticos y epigenéticos en el cromosoma 11p15.5 en el 40% de los casos y por disomía uniparental materna del cromosoma 7 en el 10%.
Métodos: Se identificaron los pacientes con diagnóstico de síndrome de Silver-Russell del Hospital Infantil de México Federico Gómez atendidos de 1998 a 2012; se reevaluaron 20 pacientes según los criterios clínicos internacionales, y se confirmó el diagnóstico en nueve sujetos.
Resultados: Todos los pacientes presentaron restricción del crecimiento intrauterino y talla baja, ambos criterios diagnósticos mayores. La macrocefalia relativa estuvo presente en el 78% y la asimetría corporal solo en el 33%. Otras características, como la acidosis tubular renal, estuvieron presentes en más del 50%.
Conclusiones: El diagnóstico del síndrome de Silver-Russell es complejo, por lo que contar con criterios clínicos adecuados es fundamental. Dado que la talla baja es la principal solicitud de atención médica en este síndrome, es relevante establecer diagnósticos diferenciales y valorar el crecimiento y desarrollo de todos los pacientes para identificar a aquellos en quienes la talla baja forma parte de una entidad sindrómica y que serían candidatos para realizar estudios moleculares. Este abordaje tendrá implicaciones para su manejo, pronóstico y asesoramiento genético.
1. Introduction
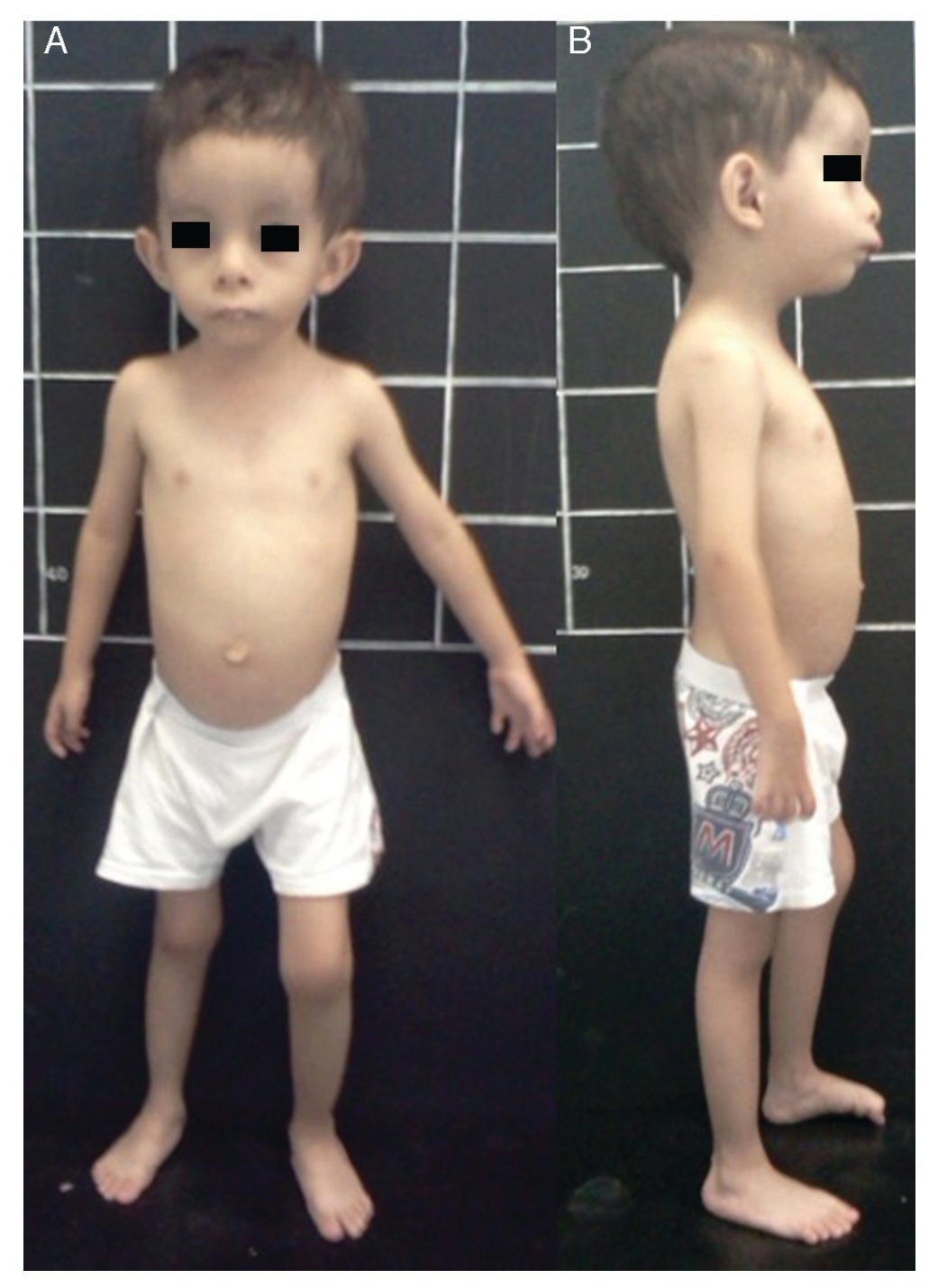
Silver-Russell syndrome (SRS) (MIM 180860) is characterized by intrauterine and postnatal growth restriction (IUGR), relative macrocephaly, triangular face, facial and/or body asymmetry and alterations in feeding (Fig. 1).1 It was described for the first time in 1953 by Henry Silver, a U.S. pediatrician who reported two children with low birth weight, postnatal growth restriction and body asymmetry.2 With no prior knowledge of this, in 1954, the English physician Alexander Russell described five similar patients.3
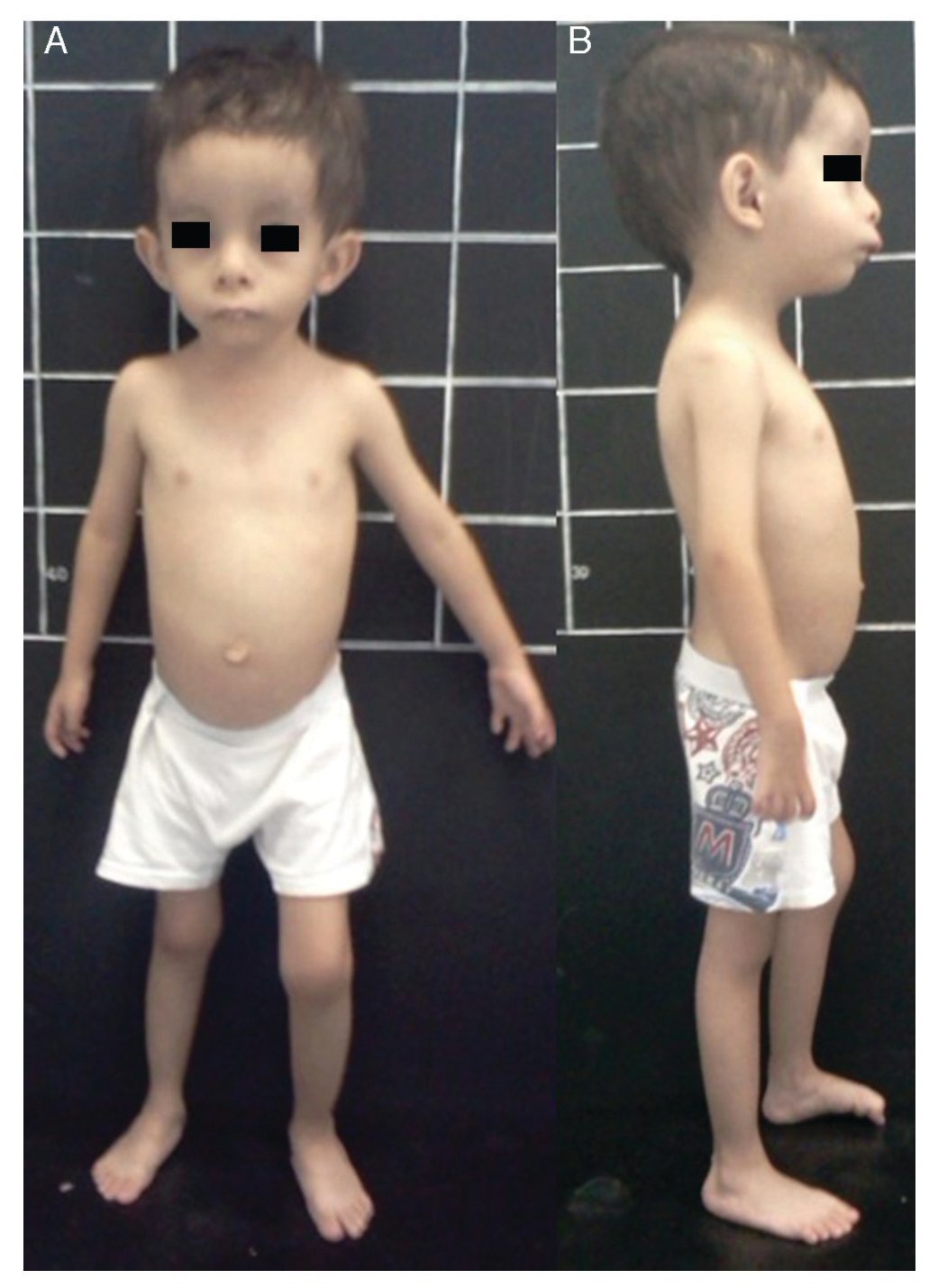
Figure 1 Phenotype of a patient with Silver-Russell syndrome (SRS). (A) Frontal view. (B) Side view. Note hemibody asymmetry, relative macrocephaly, triangular face, frontal bossing and retromicrognathia.
The incidence of SRS is 1:3000 to 1:10,000 live births and occurs in all ethnic groups without gender predilection.4 Patients with SRS, in addition to the previously mentioned clinical features, presented hypotonia and muscular hypotrophy, “cafe au lait” spots, delayed closure of fontanelles, prominent forehead, descending lip corners, micro/retrognathia, alterations in the interdental spaces, dysmorphic low-set ears, cryptorchidism, hypospadias, camptodactyly, joint contractures, clinodactyly of the fifth finger, syndactyly between the second and third toes, excessive sweating, hypoglycemia, high-pitched voice, psychomotor developmental delay and gastroesophageal reflux disease (GERD)1,4,5 (Fig. 1). There is variable expressivity observed in SRS with a clinical spectrum that can range from the classical picture to minimal clinical manifestations that may even go unnoticed. These, coupled with the difficulty of defining the principal clinical features of this disorder, make the clinical diagnosis difficult and therefore the syndrome is considered to be underdiagnosed.5
Although most cases of SRS are sporadic in presentation, involvement of genetic factors in its etiology has been demonstrated. There are reports of familial presentations suggesting different patterns of inheritance: autosomal dominant or recessive6 (and anecdotally X linked [MIM 312780]). There have also been familial cases identified associated with various chromosomal aberrations including numerical and structural rearrangements that involve in a recurrent fashion only chromosomes 7, 11p and 17q.7,8
It was initially proposed that SRS had as an etiological basis an intrauterine change or stress at 6-7 weeks of gestation.9 Other authors took into consideration both the lack of response of a target organ to growth hormone as well as a structural change in its molecule.4,9 Currently it is believed that SRS has genetic heterogeneity because several genetic and epigenetic mechanisms are involved in its etiology, principally due to changes in the imprinting.10 In the human genome a group of imprinted genes exist, i.e., genes that exhibit an expression different from its alleles depending on the progenitor from which they are inherited. The imprinted genes are involved in different aspects of growth and behavior for which several syndromes associated with disorders on the imprinting are clinically characterized according to these types of alterations.11 With regard to SRS, the mechanisms that may affect the imprinting are diverse and are associated with changes that modify the patterns of methylation in the 11p15.5 region in 40-50% of cases and a maternal uniparental disomy of chromosome 7, UPD(7)mat, in 10%.1,12
Genomic imprinting is an epigenetic phenomenon whereby the maternal and paternal germinal lines give rise to differential markings to groups of genes located in specific chromosome regions that are inherited in the gametes. Epigenetic markers include differences in methylation of CpG dinucleotides located in regulatory sequences of the imprinted genes or chromosomal regions referred to as imprinting control regions (ICR) and histone modifications that change the structure of the chromatin. These modifications are stable during mitotic division and are maintained during growth and development of the individual. Epigenetic differences between maternal and paternal copies of imprinted alleles permit gene expression of a single allele while suppressing the expression of the other, and alterations in the establishment or maintenance of the imprinting can lead to an overexpression or failure of the products of the genes involved.13
The term uniparental disomy (UPD) refers to the presence of both homologous chromosomes inherited from the same parent. UPD could present two variants: 1) isodisomy when only one of the homologous chromosomes is duplicated and 2) heterodisomy when both homologous chromosomes are inherited from one parent. The UPD can be caused by different mechanisms, among them: 1) monosomic rescue, either by fertilization of a nullisomic gamete or by an error in mitotic disjunction; 2) by events of mitotic recombination, 3) trisomic rescue, or 4) gamete complementation. The UPD of chromosomes or regions with imprinted genes modifies the functional genetic dose causing disorders of growth and development.14
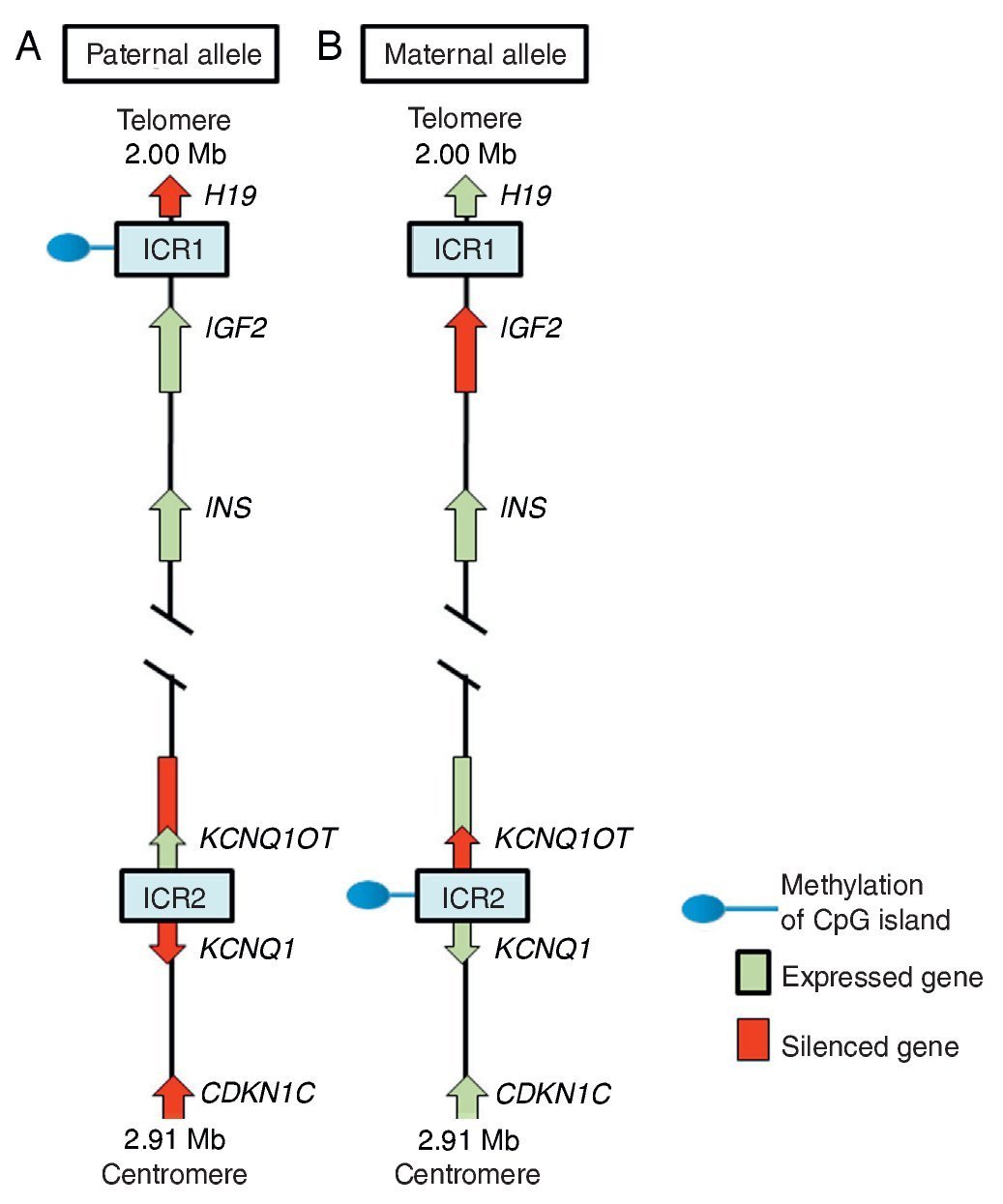
In region 11p15.5 there is a cluster of imprinted genes relevant for SRS12 (Fig. 2), which play an important role in the control of fetal growth.15 This chromosomal region is organized in two different domains or imprinted regions, each under the control of its own ICR, which acts in cis, i.e., that regulates the expression of the adjacent genes. ICR of the 11p15.5 region are known as ICR1 or telomeric and ICR2 or centromeric and have different regulatory mechanisms. ICR1 functions as an insulator, whereas ICR2 is the promoter of a macro-non-coding RNA (ncRNA).16 These ICR have opposed methylation patterns; ICR1 is found methylated in male gametes, whereas ICR2 is methylated in the germinal maternal line1 (Fig. 2). ICR1 controls the monoallelic expression of two widely studied genes: IGF2 that is expressed on the paternal allele and encodes for the insulin-like growth factor 2 (IGF2) and H19 expressed on the maternal allele and encodes for a long intergenic non-coding RNA (lincRNA).17 The genes H19 and IGF2 are widely expressed during embryonic development decreasing in almost all postnatal tissues. IGF2 plays a principal role as a promotor of placental and embryonic growth, whereas the exact function of H19 is uncertain. The expression of H19 and IGF2 is regulated by the binding of the CTCF protein in the hypomethylated ICR1 of the maternal allele. Cohesin proteins contribute to the genetic regulation of the binding sites of CTCF in ICR1 by the formation of chromatin loops that allow the expression of one or the other gene.16
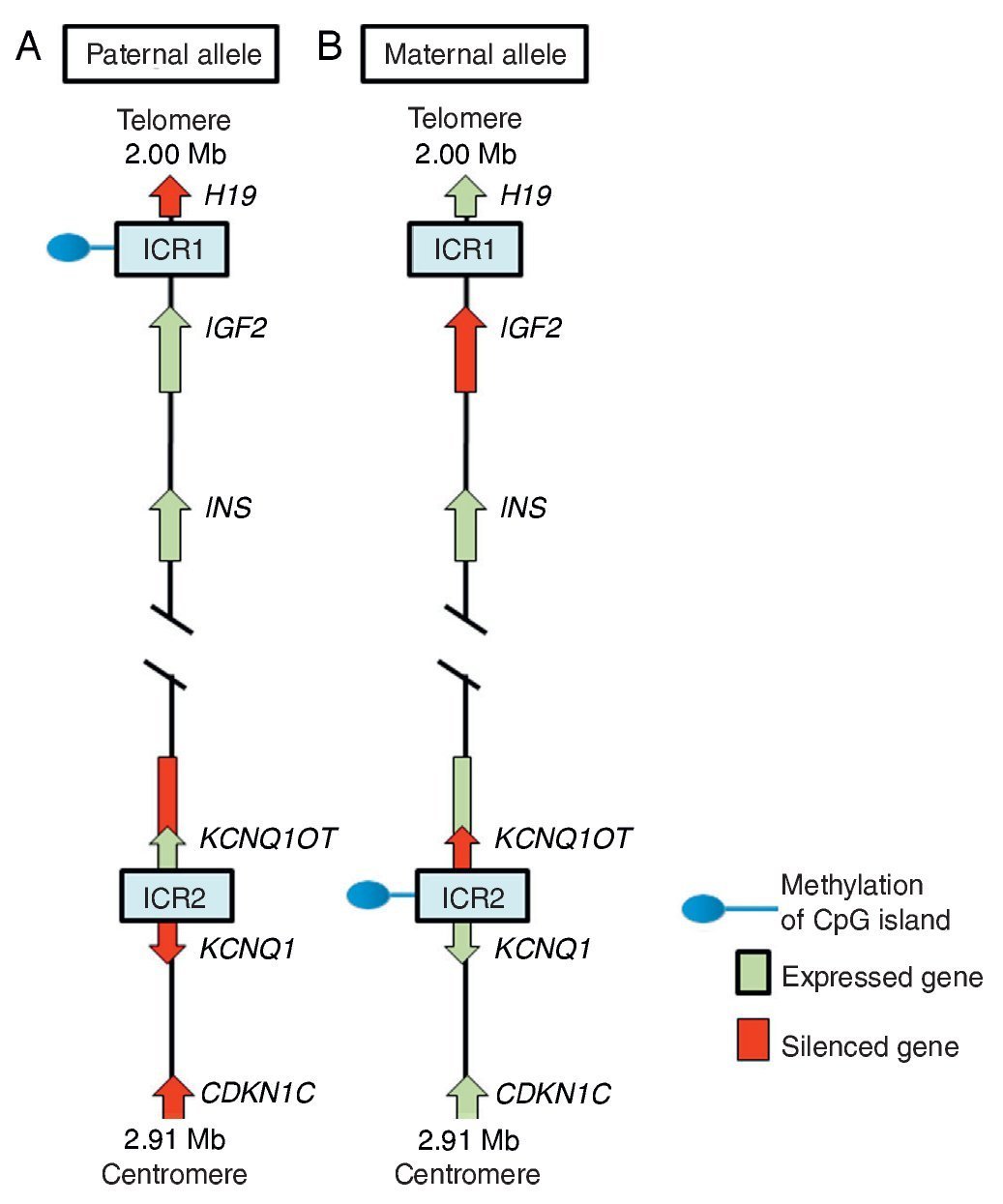
Figure 2 Imprinted region in chromosome 11p15.5. (A) Paternal chromosome and (B) maternal chromosome showing differences in methylation patterns of imprinting control regions (ICR1 and ICR2) and in the expression of the alleles of genes H19, IGF2, KCNQ1,KCNQ1OT and CDKN1C.
ICR2, which includes KvDMR1, regulates the expression of KCNQ1, KCNQ1OT1 and CDKN1C genes. In normal individuals, ICR2 is methylated in the maternal allele and results in the loss of expression of KCNQ1OT1 and the expression of KCNQ1 and particularly of CDKN1C, which is a negative regulator of cellular proliferation and growth participating in human fetal development as it encodes for an inhibitor of the cyclin-kinase complexes.18 RNA KCNQ1OT1 is expressed only from the paternal allele and results in the silencing of all imprinted genes in the domain including CDKN1C. In the hypomethylated ICR2 in the paternal allele, the binding of CTCF has also been identified.16
Beckwith-Wiedemann syndrome (BWS, MIM 130650) was the first disease in which changes in the imprinting of the 11p15.5 region were described. When the clinical manifestations are considered, it is particularly interesting that whereas SRS is characterized by IUGR and short stature, BWS is considered a syndrome of overgrowth.19 In 40-50% of the cases of SRS there is hypomethylation in the ICR1 of the paternal chromosome 11p15.5, which leads to an overexpression of the gene H19 and to a decrease of the expression of the growth promoter IGF2.1 In some cases SRS is due to a duplication of the maternal allele or to segmentary maternal UPD of chromosome 11 with increase in the expression of KCNQ1 and CDKN1C genes.16,18 ICR2 is rarely affected by epimutations in SRS, whereas hypomethylation of the maternal allele of this differentially methylated region is the principal cause of BWS.18,19
UPD(7)mat has been confirmed in 10% of cases with SRS and has been linked with alterations in the expression of imprinted genes on chromosome 7; therefore, candidate regions and genes are being investigated on this chromosome.8,14 In particular, imprinted genes have been identified in 7p11.2-p13 and 7q31-q34. Each of these loci contains as a minimum one candidate gene that participates in growth regulation, among them is the gene GRB10, which is expressed from the maternal allele and encodes for growth factor receptor-bound protein 10 and the PEG1/MEST gene expressed on the paternal allele and encodes for the mesoderm-specific transcript.10
The phenotype of patients with SRS shows differences depending on its etiology. Patients with hypomethylation of ICR1 of 11p15.5 have the “classic” phenotype of SRS as well as a greater incidence of body asymmetry when compared with patients whose etiology is due to a UPD(7)mat.7,20
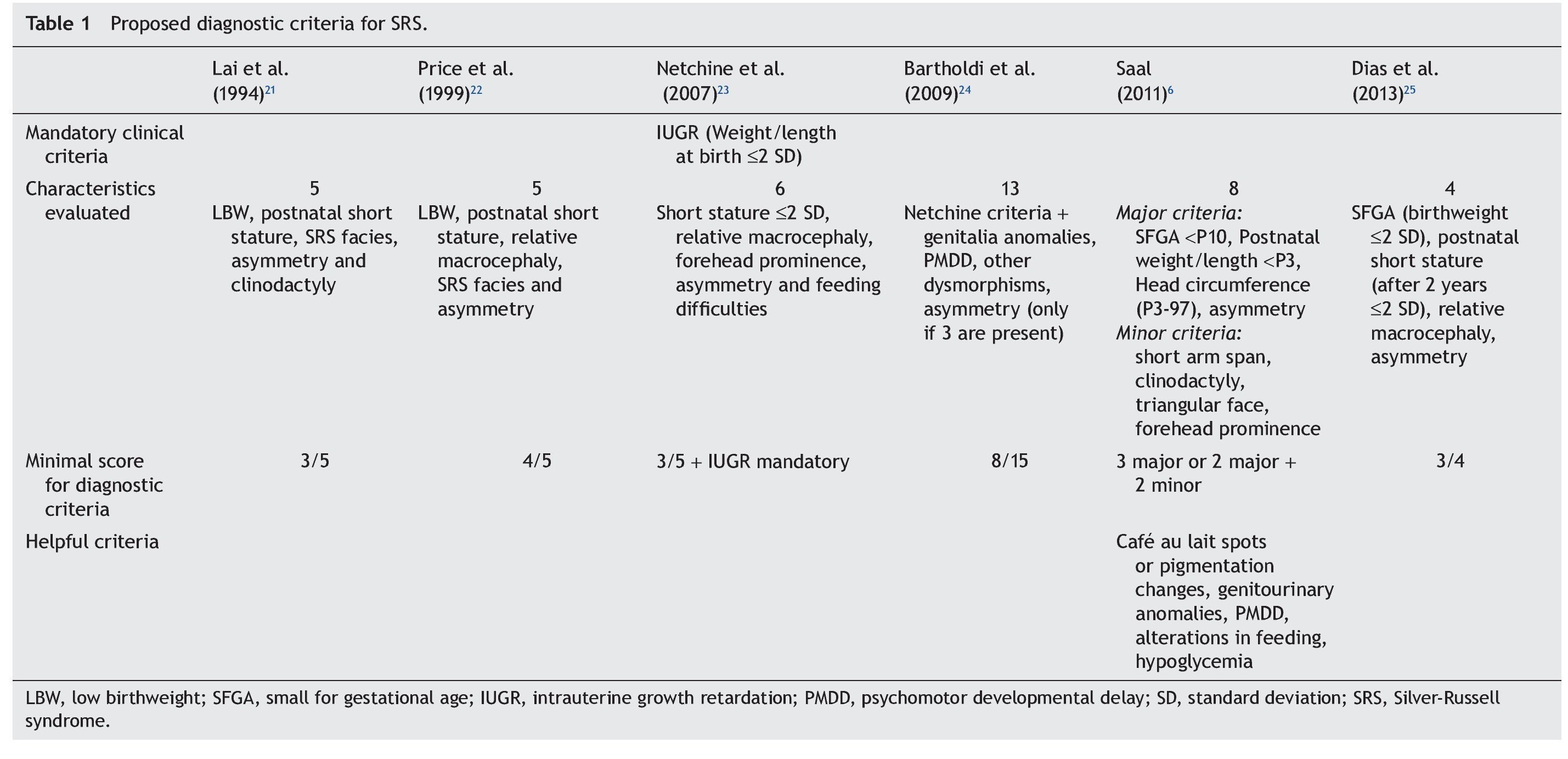
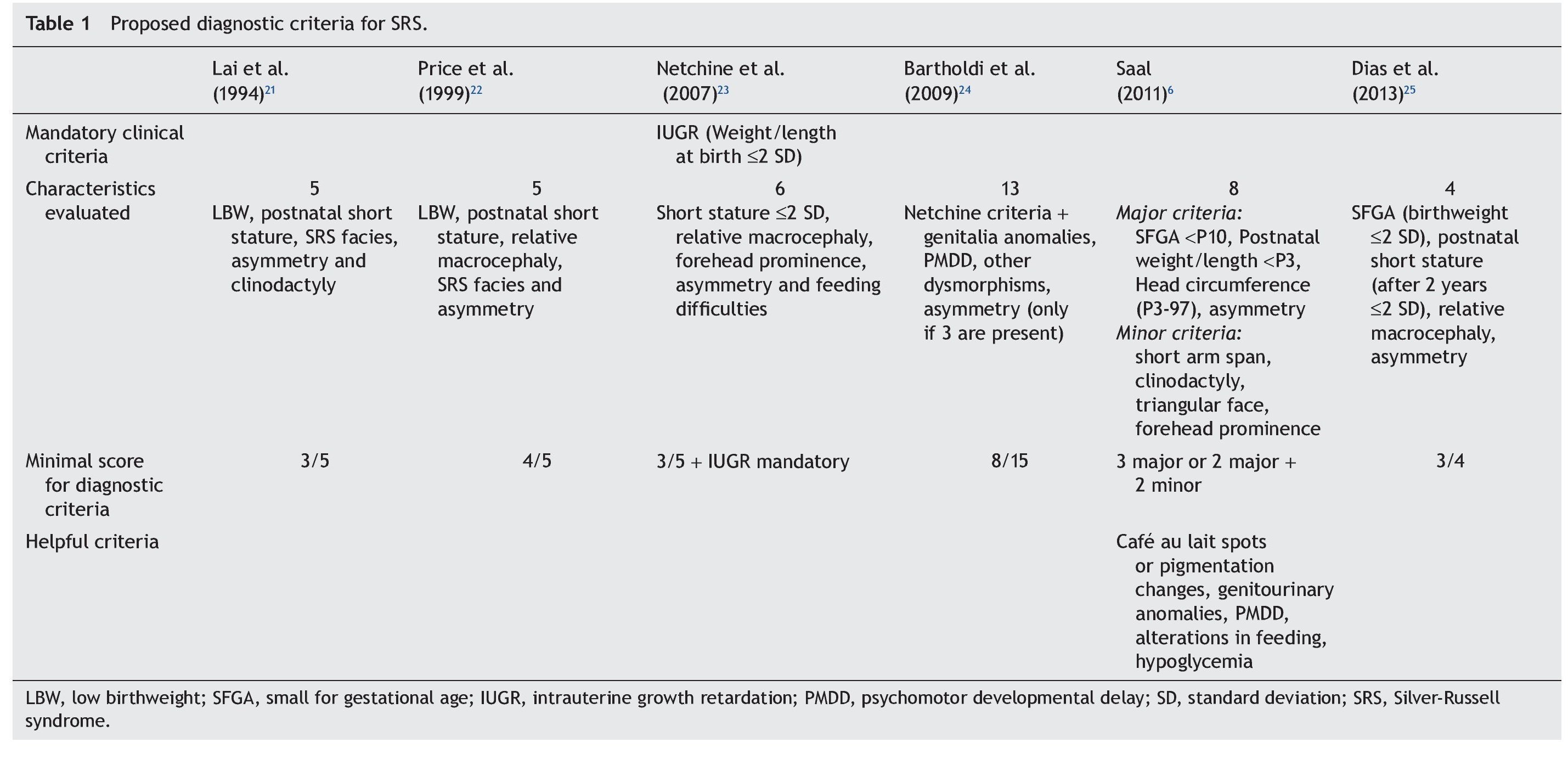
Because the etiological bases of SRS are complex, molecular diagnosis is complicated and in half of the cases is inconclusive. Therefore, it is necessary to rely on an accurate clinical diagnosis; however, this continues to be a challenge because of the variability of the phenotypic expression. To date, various evaluation systems have been proposed based on a given score to delineate the syndrome (Table 1)6,21-25 without a universally accepted scoring system or diagnostic criteria.26,27 In addition, facial characteristics tend to be attenuated during late infancy and more so in adulthood.1,25 Because of this, SRS is often considered to be a presumptive diagnosis.
The differential diagnosis of SRS includes any entity that involves IUGR and short stature and syndromes secondary to chromosomal aberrations and alterations in DNA repair as well as the 3-M [MIM 273750], Dubowitz [MIM 223370], IMAGE [MIM 614732] syndromes. Fetal alcohol syndrome should also be ruled out.9 These aspects all result in complications in providing accurate genetic counseling.
The clinical characteristics of a series of patients with clinical diagnosis of SRS managed at the Department of Genetics at the Hospital Infantil de Mexico Federico Gomez from 1998-2012 are described in this paper. This work is part of the research protocol “Implications of methylation pattern of the 11p15.5 region as an etiological mechanism of isolated hemihyperplasia” (HIM/2012/007). As previously described by Moreno et al.,28 patients with a clinical diagnosis of SRS were included in this protocol as controls for molecular study of the 11p15.5 region. In order to accomplish this, a review of the clinical record, clinical re-evaluation of the patients identified and finally a statistical analysis of the characteristics present in the patients with diagnostic criteria compatible with SRS was carried out (Table 1). Molecular study of these cases will be reported in the context of the protocol performed.
2. Methods
The study group included patients registered with a clinical diagnosis of SRS in the Department of Biostatistics and Clinical Archives of our institution from 1998 to 2012. A descriptive and retrospective study was carried out. There were 20 patients identified who were clinically reevaluated according to the criteria proposed by Saal6 to confirm or rule out the clinical diagnosis of SRS. The Saal6 criteria are based on the evaluation of the presence of eight clinical characteristics divided into four major and four minor criteria, with the diagnosis being made based on the presence of three major or two major and two minor criteria. The scale also analyzes five other clinical characteristics that, if present, would further support the diagnosis (Table 1). Patients who did not meet these criteria were excluded from this analysis.
3. Results
Of the total of 20 patients identified by the registry with SRS, only nine met the clinical diagnostic criteria proposed by Saal6 for SRS. Of the 11 patients who were excluded, three were diagnosed as familial short stature, three as short stature secondary to renal tubular acidosis (RTA) and one as Dubowitz Syndrome. Four remaining patients are still under study.
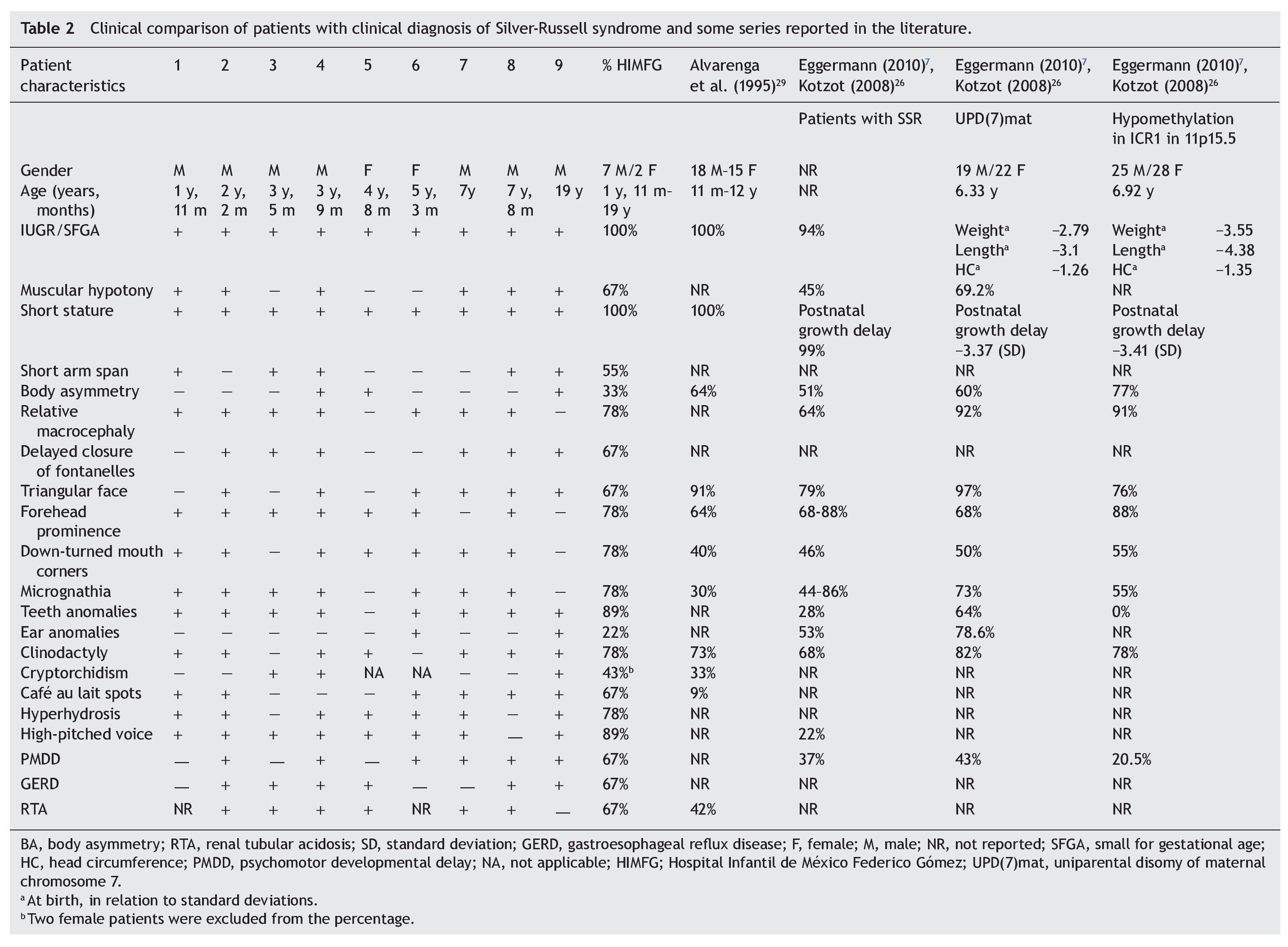
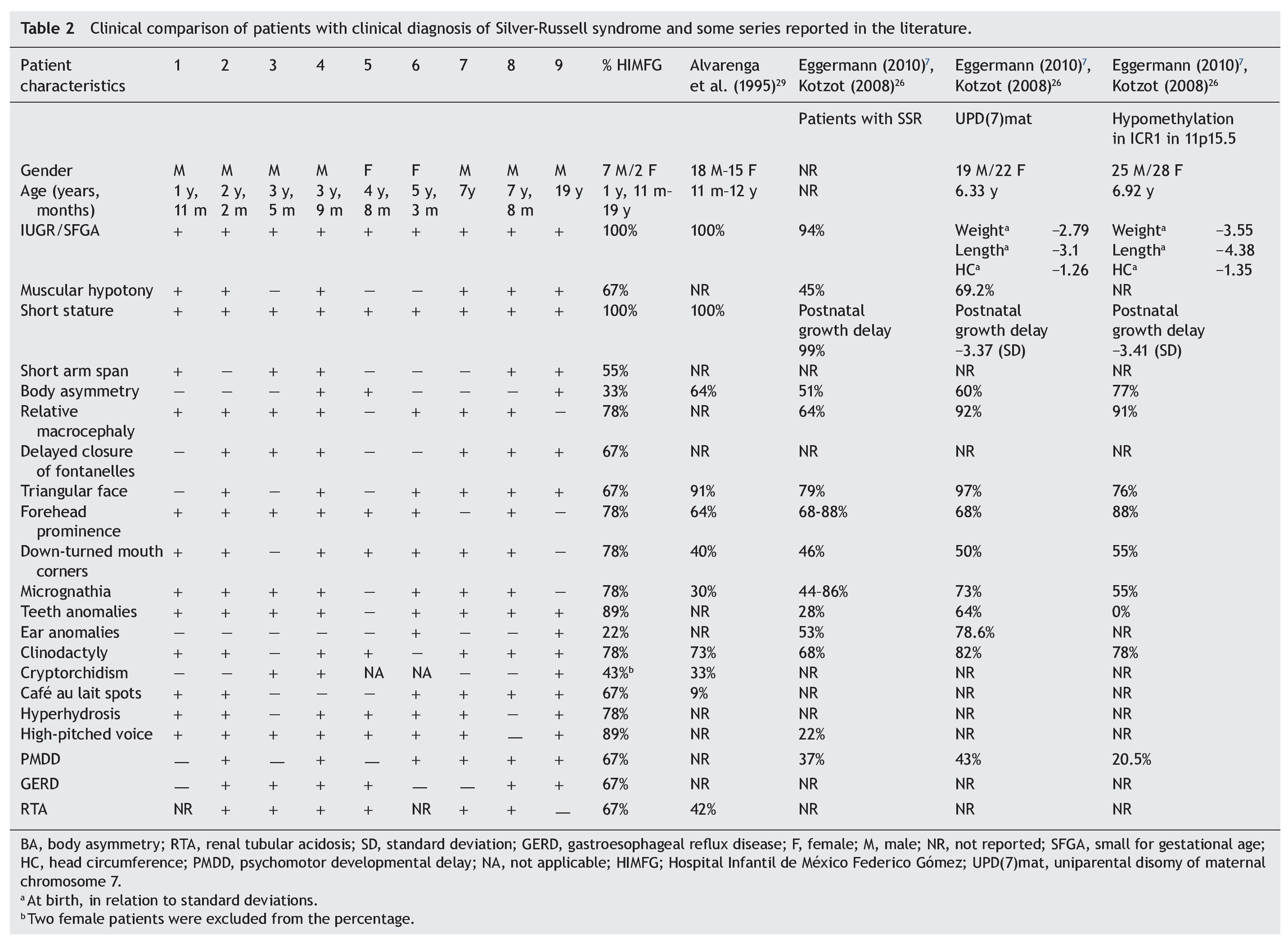
With regard to the analysis of the clinical characteristics of the nine patients with clinical diagnosis of SRS, seven were males and two females, with an age range between 1 year 11 months and 19 years of age. The karyotype was normal in five patients (patients 2, 4, 6, 7 and 9) who were cytogenetically studied. Individual clinical characteristics of the nine patients analyzed and the percentages in which they were found in this series are shown in Table 2. 26,27,29
4. Discussion
Evaluation of 20 patients registered with a diagnosis of SRS using the Saal6 criteria allowed us to rule out the diagnosis in 11 of the patients and in 7/11 patients (64%) a different diagnosis was made. Of the nine patients (45% of total registered) in whom the diagnosis of SRS was confirmed, there was a higher percentage of male patients (77.77%). These data are striking because it is considered that SRS has no predilection for gender.4 In this series, 43% of male patients had uni- or bilateral cryptorchidism (Table 2). In the series by Alvarenga et al.,29 cryptorchidism was present in 33% of their patients. The high frequency of this characteristic could influence the early care of a greater number of male patients with SRS because it is a condition of alarm for surgical care, regardless of whether other data from SRS are present.
SRS is a disorder that is difficult to diagnose because of the wide range in its expression and the low specificity of its principal manifestations. Short stature is the cardinal sign for which most patients with SRS seek medical care. In our series, all patients with confirmation of the diagnosis had short stature. This information is equally important to consider the differential diagnosis with phenotypic similarity. Short stature is defined as that which is <2 standard deviations (SD) from the mean for age.27,30 The most common causes are familial short stature and constitutional growth delay which, together, add up to 75%. Other etiologies are chronic diseases (10%), syndromic disorders (6%), chromosomal abnormalities (5%), skeletal dysplasias (1%) and growth hormone deficiency or receptor insensitivity (1-2%) as well as an inadequate intake of nutrients and psychological problems.31 Therefore, it is important to rule out those causes that are much more common before considering the diagnosis of SRS.
In patients with short stature of unknown cause, a strict somatometry of the body segments should be performed in order to assess the growth rate and compare the height of the patient with the target family height (TFH).30 Electrolyte levels, urea concentration, thyroid function, complete blood count, urinalysis, anti-gliadin antibodies, bone age and additional endocrinological tests required according to the case should also be analyzed. Cytogenetic analysis should also be considered, for example, to rule out Turner syndrome or in patients with mental retardation with or without malformations or dysmorphias. In cases of TFH, bone maturation is consistent with chronological age and final height should be estimated with the TFH. In cases with constitutional growth and developmental delay, bone maturation is consistent with the age for height and the prognosis for normal adult height is good.30-32 Short stature is proportional, although there can be body asymmetry and, in fact, IUGR, which is manifested as lower than expected height at birth. This pattern is maintained during subsequent development <2 SD.
Among the principal syndromes associated with short stature with a history of low birth weight is Dubowitz Syndrome, an autosomal recessive disorder. There have been ∼200 cases described and the syndrome has no ethnic or gender predilection.33 This disorder is the main differential diagnosis of SRS because the two conditions share several clinical features such as IUGR, postnatal growth retardation, micrognathia, dental crowding, abnormal ears, genitourinary abnormalities, and high-pitched voice. They also have triangular face, facial asymmetry, and ptosis (65%).33,34 In this study, in one patient in whom SRS was ruled out, the diagnosis of Dubowitz Syndrome was made because the patient met only two major criteria for SRS (low weight for gestational age and low postnatal weight and stature Three of the patients excluded from this series had short stature not less than P3 and secondary to RTA diagnosed. RTA alone can be a cause of non-syndromic short stature. No additional data associated with SRS were found in these patients. It is notable that RTA was present in 67% of patients in this series and in 42% of patients described by Alvarenga et al.,29 raising the consideration of whether or not RTA is a characteristic of SRS, as until now this is not one of its diagnostic criteria. The percentages of the clinical characteristics identified in our group of patients with SRS were consistent with the published data.5,7,29 (Table 2). As already mentioned, all patients had IUGR and short stature, cardinal clinical data on all proposed diagnostic criteria (Table 1). Using the proposal by Saal,6 all patients met at least three major criteria and only patient 4 presented the four major criteria. Of the other major criteria, 78% of our patients had relative macrocephaly and only 33% body asymmetry in contrast to what has been reported in other series (64 and 51%, respectively).5,7,26 It was notable that half of our patients demonstrated short arm span, although this is a minor criterion. It is not reported in percentages in other studies, but its high frequency in our population would support its use as a major criterion for SRS. The high frequency of the characteristics identified as minor criteria by Saal6 both in our population as well as in those reported in the literature would support the use of these characteristics in the diagnosis of SRS.5,7,26 Among the supporting diagnostic criteria demonstrating a high frequency of presentation in our patients are cafe au lait spots, GERD, psychomotor developmental delay and genitourinary abnormalities (Table 2). Additionally, other features present were high-pitched voice and teeth abnormalities, down-turned lip corners, micrognathia, hyperhydrosis, muscular hypotonia and delayed closure of the fontanelles, all at a higher rate than previously reported. Ear abnormalities were found in a lower proportion than what has been reported (Table 2). Even though our series was small, it allowed us to corroborate that by using the clinical criteria proposed by Saal6 it is possible to have a better approximation of the clinical diagnosis of SRS. When the criteria by Dias et al.25 were taken into consideration, 8/9 patients met 3/4 of the diagnostic characteristics, whereas the remaining patient met all of the diagnostic characteristics. With the remainder of the proposed criteria including those of Bartholdi et al.,24 which are those that use the greatest number of clinical characteristics, diagnosis of SRS is also confirmed in the nine cases. The 11 cases in whom the diagnosis was excluded using the Saal6 criteria also did not meet the diagnostic criteria used by the other evaluation scales. Diagnosis of SRS is fundamentally based on clinical criteria even though there are several molecular causes known. In up to 50% of the patients with a clinical diagnosis, the etiology is not identified. In those cases in which the molecular alteration of the SRS is determined, it is possible to establish a correlation with the clinical characteristics and provide genetic counseling with a molecular basis. Thus, it has been reported that in cases with 11p15.5 epimutation, the clinical picture most often presented is body asymmetry and frontal bossing when compared to those with UPD(7)mat or other causes.1,4,5,7,26 On the contrary, facial alterations such as triangular face, frontal bossing, micrognathia, dental and ear abnormalities, muscular hypotonia and PMDD are prominent in patients with UDP(7)mat.1,4,5,7,26 The most common causes identified in SRS are the epimutations. In our series only patients 4, 5 and 9 had body asymmetry (Table 2) and may correspond to this etiology. However, we must take into consideration that if the application of diagnostic clinical criteria is very strict, one may run the risk of ruling out patients who have a more subtle phenotype as suggested by Eggermann.7 With regard to genetic counseling, when there is a clinical diagnosis of SRS due to an imprinting defect in the ICR1, the risk of this occurring in other family members is rarely found increased in relation to the general population. When the etiology is due to UPD(7)mat the risk is the same as that for the general population.6 However, when dealing with a duplication or mutation of the ICR2 on the maternal chromosome 11p15.5, the risk of recurrence is 50% when there is maternal transmission.35 In this report of patients with SRS that gathers the experience of one of the national pediatric reference centers during the period between 1998 and 2012, the diagnostic complexity of SRS based only on clinical criteria is reflected. To date, there is no accepted international index for its diagnosis; however, a widely considered option is the criteria proposed by Saal,6 which was applied in our patients. This analysis demonstrated that when strict criteria are used, only 45% of the patients in whom the diagnosis of SRS was initially considered did correspond to this disorder. This fact reflects the importance for the general practitioner and the pediatrician to carry out strict growth and developmental evaluation of all their patients during different stages and to be mindful of the evaluation of small dysmorphias or of the criteria that support the diagnosis, in particular in patients studied due to short stature. This approach will allow ruling out other causes and identifying those patients who may benefit from molecular studies to rule out syndromatic disorders such as the one we are discussing, which would have important implications for the management, prognosis and genetic counseling. Funding Research protocol “Implications of the methylation pattern in the 11p15.5 region as etiologic mechanism of the isolated hemihyperplasia” (HIM/2012/007), Federal funding. Conflict of interest The authors declare no conflict of interest of any nature. Received 10 March 2014; * Corresponding author.
accepted 9 July 2014
E-mail: vfmoran@himfg.edu.mx (V.F. Morán-Barroso).