




Hemophagocytic syndrome, macrophage activation syndrome, reactive histiocytosis or hemophagocytic lymphohistiocytosis (HLH) represent a group of diseases whose common thread is reactive or neoplastic mononuclear phagocytic system cells and dendritic cell proliferation.
Clinical caseWe present a case of an HLH probably associated with Epstein-Barr virus (EBV) in a 4-year-old male patient treated with HLH-04 protocol. Viral etiology in HLH is well accepted. In this case, clinical picture of HLH was assumed secondary to EBV infection because IgM serology at the time of clinical presentation was the only positive factor in the viral panel.
ConclusionsDiagnosis of HLH is the critical first step to successful treatment. The earlier it is identified, the less the tissue damage and reduced risk of multiple organ failure, which favors treatment response.
El síndrome hemofagocítico, síndrome de activación de macrófagos, histiocitosis reactiva o linfohistiocitosis hemofagocítica (HLH) representan un grupo de enfermedades cuyo factor común es la proliferación reactiva o neoplásica de las células mononucleares fagocíticas y del sistema de células dendríticas.
Caso clínicoSe presenta un caso de HLH sugestivo de tener una asociación con el virus del Epstein Barr (VEB) de un paciente masculino de 4 años de edad, tratado con el protocolo HLH-04. La etiología viral en HLH es reconocida. En este caso se asumió un cuadro de HLH secundario a una infección por VEB, ya que la serología de IgM en el momento de la presentación clínica fue la única positiva en el panel viral.
ConclusionesEl diagnóstico de la HLH es el primer paso crítico para el éxito del tratamiento. Entre más temprano se identifique, existe menor daño tisular y menor riesgo de falla orgánica múltiple, lo que favorece la respuesta al tratamiento.
Hemophagocytic syndrome, also known as macrophage activation syndrome, reactive histiocytosis or hemophagocytic lymphohistiocytosis (HLH), represents a group of diseases whose common factor is reactive or neoplastic proliferation of phagocytic mononuclear cells and of the dendritic cell system.1 The International Histiocytosis Society coined the term to refer to a genetic disease with severe systemic inflammation,2 which occurs as the result of a recessive genetic disorder associated with either defects in perforin or chromosome X-linked. The disease affects the communication of the innate and adaptive immune response, causing a lack of removal of the inflammatory cells harmful to the body and that should be removed once the inflammatory process has concluded. It is estimated that there are ~1.2 cases per million/year. These are uncommon cases, but its diagnosis is important because the clinical pictures are very aggressive and evolve in a fulminant manner and with multiple organ failure if a precise and timely diagnosis is not carried out.3 In this disease, T and B lymphocyte count and circulating monocytes show normal values. However, the defect is found in the function of T-lymphocytes and natural killer cells (NK) as well as in the activation of CD4+ and CD8+ T lymphocytes.4 Other altered functions are the response of lymphocytes to mitogen activation, oxidation of glucose in phagocytes and, in some cases, decrease of IgA5 antibodies. Clinical onset may be spontaneous. Prior reports described that ~17% of the cases are associated with viral infections, among these, the Epstein-Barr (EBV) virus.6 This report presents a case of HLH suggested to be associated with EBV.
2Clinical caseWe present the case of a 4-year-old male patient. The patient’s mother was 33 years of age, healthy and denied any drug addictions. The father was 35 years of age with a secondary school education. He was apparently healthy and admitted occasional smoking. There is an apparently healthy 11-year-old brother. The patient is originally from and resident of Ecatepec, State of Mexico. The family lives in their own home with all urban services. Overcrowding or coexistence with animals was denied. The patient demonstrated adequate hygiene and dietary habits. Vaccination schedule was incomplete because there was a lack of DPT booster vaccine. The patient showed appropriate psychomotor development although he did not attend any child development center (Centro de Desarrollo Infantil, CENDI). The patient was the product of a GII with regular prenatal control and the mother received iron and folic acid intake from the first trimester without complications. The patient was born by cesarean section at 39 weeks of gestation due to nuchal cord. Usual resuscitation maneuvers were carried out. Apgar score was 8/9. The infant cried and breathed at birth. Birthweight was 3150g and length was 54cm. The infant was discharged without complications.
In March 2014, the patient began with right earache. He was seen in consultation by a private physician. Treatment with acetaminophen, ambroxol and amoxicillin was prescribed for 7 days, without improvement. He also began with fever up to 38.9°C. Treatment with trimethoprim with sulfamethoxazole was added due to the persistence of fever. Complete blood count was done. Parameters low for age were seen for hemoglobin, hematocrit and platelets: hemoglobin 10.5g/dl (reference value: 11.60–15.30g/dl), hematocrit 31.7% (reference value: 35.6–46.52%), leukocytes 4,920/μl (reference value: 4,290–12400/μl, neutrophils 1082/μl (reference value: 983–3231/μl), bands 27%, atypical lymphocytes 5%, platelets 97,000/μl (reference value 147–384,000/μl).7 Physical examination observed retroauricular and inguinal lymph nodes, hepatosplenomegaly, and persistent fever. The patient was referred to hematology for study protocol with diagnosis of lymphoproliferative syndrome.
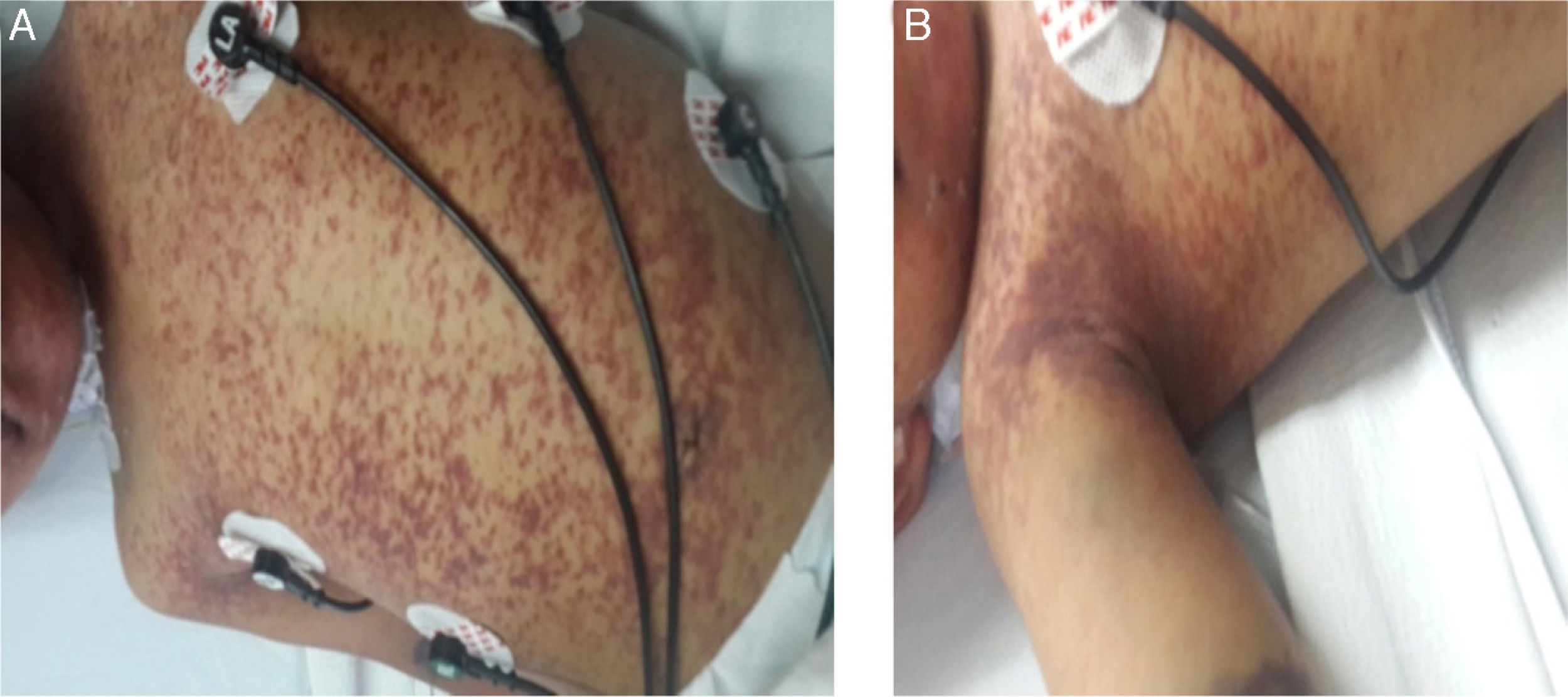
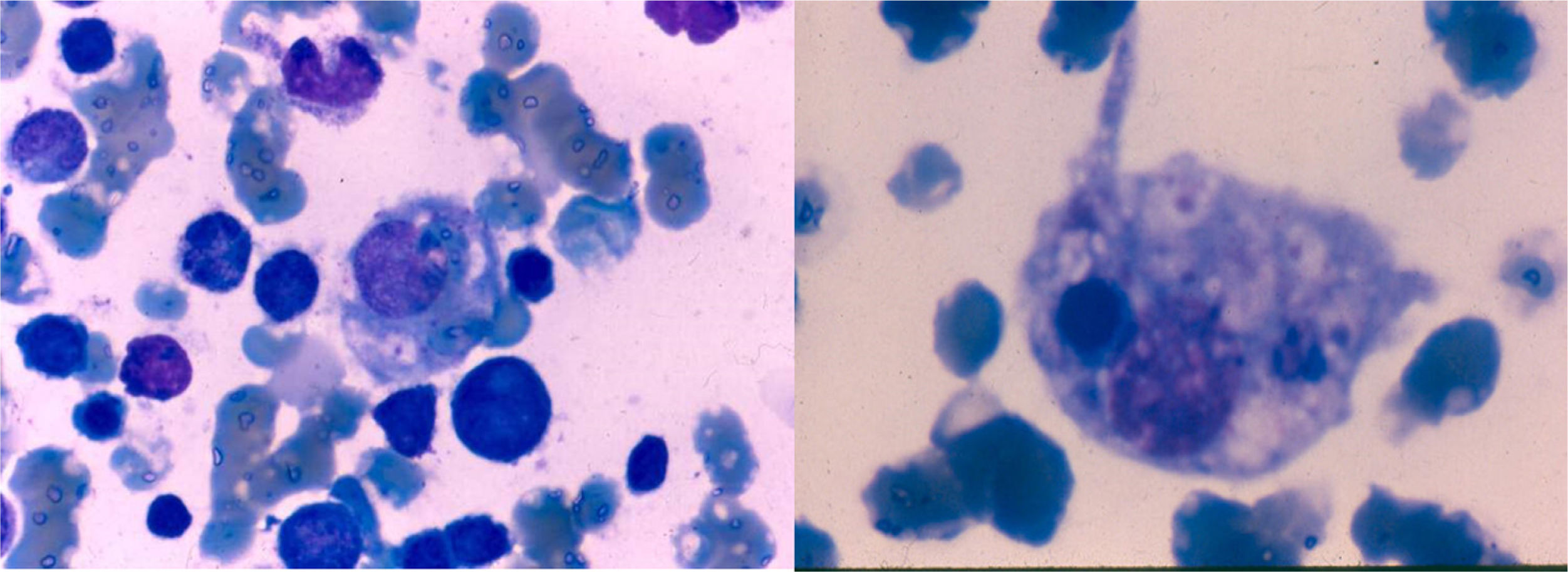
The patient was observed with pallor, adequate hydration and was active, reactive, and neurologically intact with generalized erythematous violaceous rash, ecchymosis at the venopuncture sites (Fig. 1 A and 1B), congestive oropharynx without exudate, and neck with mobile bilateral adenomegalies of 1.5cm diameter. Pulmonary fields were well ventilated and he was tachycardic. Abdomen was painful on deep palpation, with hepatomegaly at 4-5-6cm below the costal margin and splenomegaly at 6cm and inguinal regions with 1.5cm mobile adenomegalies. Genitalia were consistent with age and gender and extremities were intact with 3-sec capillary refill. Bone marrow aspirate did not show the presence of blasts and there were abundant histiocytes with hemophagocytosis (Fig. 2).
 Disseminated erythematous violaceous rash and visceromegalies.")

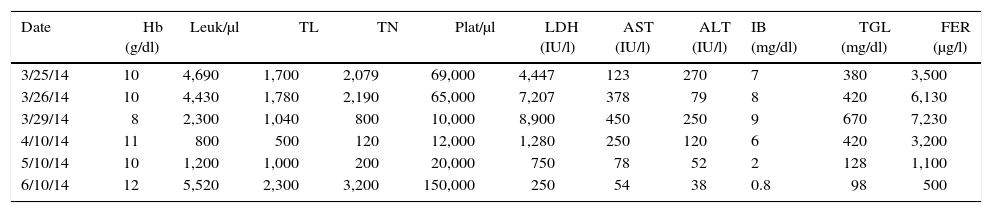
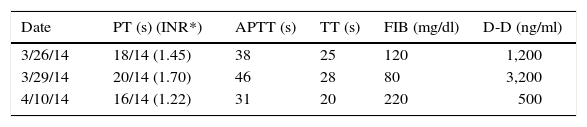
The following day the patient was lethargic with persistent fever up to 40°C and data of multiple organ failure. He subsequently presented respiratory deterioration that warranted endotracheal intubation through rapid sequence intubation with cardiac arrest, which reverted with a single cycle of compressions. He was admitted to the Pediatric Intensive Care Unit (PICU). The same day he began treatment protocol HLH-04.8 Briefly, the therapy was carried out during the first 8 weeks with etoposide, dexamethasone and cyclosporin A, without intrathecal chemotherapy because the cerebral spinal fluid was acellular and clinically without neurological alterations. He remained for 2 days in the PICU and continued being managed in the Pediatric Hematology unit with satisfactory progress. It is important to mention that care support measures were very important; among these, blood components were all irradiated with white blood cells reduction filter. Two weeks later his hematological parameters, liver function tests and ferritin were normalized (Tables 1 and 2). At the same time the IgM EBV-positive serology was confirmed with a viral panel (Table 3) as well as changes in the lymphocytic subpopulations (Table 4). He completed the treatment scheme as mandated in the HLH-04 protocol.7 He is currently disease-free and is being followed-up as an outpatient.
Hematologic parameters, hepatic function and ferritin.
| Date | Hb (g/dl) | Leuk/μl | TL | TN | Plat/μl | LDH (IU/l) | AST (IU/l) | ALT (IU/l) | IB (mg/dl) | TGL (mg/dl) | FER (μg/l) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3/25/14 | 10 | 4,690 | 1,700 | 2,079 | 69,000 | 4,447 | 123 | 270 | 7 | 380 | 3,500 |
| 3/26/14 | 10 | 4,430 | 1,780 | 2,190 | 65,000 | 7,207 | 378 | 79 | 8 | 420 | 6,130 |
| 3/29/14 | 8 | 2,300 | 1,040 | 800 | 10,000 | 8,900 | 450 | 250 | 9 | 670 | 7,230 |
| 4/10/14 | 11 | 800 | 500 | 120 | 12,000 | 1,280 | 250 | 120 | 6 | 420 | 3,200 |
| 5/10/14 | 10 | 1,200 | 1,000 | 200 | 20,000 | 750 | 78 | 52 | 2 | 128 | 1,100 |
| 6/10/14 | 12 | 5,520 | 2,300 | 3,200 | 150,000 | 250 | 54 | 38 | 0.8 | 98 | 500 |
Hb, hemoglobin; Leuk, leukocytes; TL, total lymphocytes; TN, total neutrophils; Plat, platelets; LDH, lactate dehydrogenase; AST, aspartate aminotransferase; ALT, alanine aminotransferase; IB, indirect bilirubin; TGL, triglycerides; FER, ferritin.
Coagulation tests.
| Date | PT (s) (INR*) | APTT (s) | TT (s) | FIB (mg/dl) | D-D (ng/ml) |
|---|---|---|---|---|---|
| 3/26/14 | 18/14 (1.45) | 38 | 25 | 120 | 1,200 |
| 3/29/14 | 20/14 (1.70) | 46 | 28 | 80 | 3,200 |
| 4/10/14 | 16/14 (1.22) | 31 | 20 | 220 | 500 |
PT, prothrombin time; APTT, activated partial thromboplastin time; TT, thrombin time; FIB, fibrinogen; D-D, D-dimer; INR, internationalized normal ratio (reference values 0.9–1.3).
Viral serology of the patient at diagnosis.
| Date | HBVs | HBVc | HCV | HIV | EBV (VCA) IgG | EBV (VCA) IgM | TOXO IgG/IgM | RB IgG/IgM | CMV IgG/IgM | HSV 1 + 2 IgG/IgM |
|---|---|---|---|---|---|---|---|---|---|---|
| 3/29/14 | − | − | − | − | + | + | −/− | +/− | −/− | −/− |
HBVs, antigen-soluble hepatitis B virus; HBVc, hepatitis B virus antigen core; HCV: hepatitis C virus; HIV, human immunodeficiency virus; EBV, Epstein-Barr virus; VCA, viral capsid antigen; TOXO, toxoplasma; RB, rubeola; CMV, cytomegalovirus; HSV 1 + 2, herpes simplex virus 1 and 2.
Subpopulations of lymphocytes and immunoglobulins.
| Date | Leu/μl | CD3+ | CD4+ | CD8+ | CD4+/CD8+ | NK | NKT | IgA | IgM | IgG |
|---|---|---|---|---|---|---|---|---|---|---|
| 3/30/14 | 1,540 | 882 (66%) | 454 (34%) | 375 (28%) | 1.2 | 41.53 | 11 | 193 | 115 | 630 |
Leu, Leucocytes; CD, cluster of differentiation; NK, natural killer; NKT, natural killer T; Ig, Immunoglobulin.
Reference values: CD3 + : 900–4500 (43–76%); CD4 + : 500–2300 (23–48%); CD8 + : 300–1600 (14–33%); CD4+/CD8 + : 0.9–2.9; NK: 3.15%; NKT: 5–26%; IgA: 25–154; IgM: 43–196; IgG: 463–1,236.
Pathogenesis of secondary HLH is unclear although it has been associated with heterozygocity or gene polymorphisms as in the familial HLH. EBV has been one of the etiologic agents involved.9 Clinical manifestations of HLH in this patient correlated with what has been previously reported and are due to three main causes: 1): hyperactivation of the T CD8 + lymphocytes and macrophages; 2) proliferation, ectopic migration and infiltration of these cells in various organs; and 3) hypercytokinemia, with persistently elevated levels of proinflammatory cytokines resulting in progressive organ failure which can lead to death.9 These interrelated factors lead to clinical manifestations such as prolonged fever, lymphadenopathy, hepatosplenomegaly, bleeding, skin rash, central nervous system abnormalities, jaundice and laboratory findings of bicytopenia or pancytopenia, coagulopathy, hyperlipidemia, hypofibrinogenemia, hyperferritinemia, transaminasemia, hyperbilirubinemia and hyponatremia.9
Viral etiology in HLH is recognized. In this case a picture of HLH secondary to an EBV infection was assumed because the VCA (viral-capsid antigen) serology IgM at the time of the clinical presentation was the only positive finding in the viral panel. In addition, there are results that place in evidence the use of the VCA IgM marker in patients with hemophagocytosis when the cut-off values of the test are sufficient to be considered positive.10
It was noted that 15% of patients with EBV-associated HLH develop disorders such as selective hypogammaglobulinemia of IgA or hyper-IgM syndrome. Such syndromes were not observed in this patient and, in fact, the IgA was noted to be increased. A more recently discovered mechanism in patients infected with mycobacterium describes the participation of a signaling system known as SLAM-SAP (signaling lymphocyte activation molecule) present in the membrane of B and T lymphocytes. SLAM-SAP triggers intracellular signals with several responses; among these, a discharge in IgA production, such as might have been in this case.11 This work group previously reported another case, but it was associated with cytomegalovirus in a bone marrow transplant patient.12
Diagnosis of HLH is the first critical step for treatment success. The earlier it is begun, the less tissue damage and less risk of multiple organ failure, which favors the response to the HLH-04 treatment.13,14 Participation of the PICU and the supportive care provided play a fundamental role.
Ethical disclosureProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflict of interestThe authors declare no conflicts of interest.
The authors extend their appreciation to the patients and families for their help and understanding during the treatment protocol, as well as to the personnel who participated during the diagnosis, treatment and follow-up.