



In primary immunodeficiencies there is a failure in the anti-tumor defense. Common variable immunodeficiency (CVID) is one of the most common primary immunodeficiencies characterized by an alteration in the differentiation of B lymphocytes (BL). Epstein-Barr virus (EBV) is an ubiquitous virus that selectively infects the BL. In patients with immunodeficiency, uncontrolled proliferation of infected BL and the action of viral proteins promote the development of lymphomas.
Clinical casesAt the University Hospital Sant Joan de Deu, Barcelona, 28 patients were diagnosed with CVID from 2000 to 2013. This paper describes four patients who developed non-Hodgkin’s lymphoma (NHL). The lymphoma was associated with EBV in two of the cases. Patients were < 18 years old, diagnosed with lymphoma between 4 and 13 years old. Two patients were treated with rituximab as monotherapy and achieved complete remission. Two patients were treated with CHOP (cyclophosphamide, doxorubicin, vincristine and prednisolone) and radiotherapy or rituximab and achieved complete remission.
ConclusionsEarly detection of EBV infections and NHL in all patients diagnosed with CVID is recommended, regardless of age at diagnosis.
En las inmunodeficiencias primarias existe un fallo en la defensa antitumoral. La inmunodeficiencia variable común (IDVC) es una de las inmunodeficiencias primarias más frecuentes. Se caracteriza por una alteración en la diferenciación de linfocitos B (LB). El virus de Epstein-Barr (EBV) es un virus ubicuo que infecta de manera selectiva los LB. En pacientes con inmunodeficiencias, la proliferación incontrolada de LB infectados y la acción de proteínas virales promueve la aparición de linfomas.
Casos clínicosEn el Hospital Universitario Sant Joan de Déu, Barcelona, se han diagnosticado 28 pacientes con IDVC del 2000 al 2013. En este trabajo se describen cuatro que desarrollaron linfoma no Hodgkin (NHL). El linfoma fue asociado a EBV en dos de ellos. Los pacientes eran menores de 18 años, con el linfoma diagnosticado entre los 4 y 13 años de edad. Dos de los pacientes fueron tratados con rituximab como monoterapia, y lograron la remisión completa. Dos fueron tratados con CHOP (ciclofosfamida, doxorrubicina, vincristina y prednisolona) y radioterapia o rituximab y también alcanzaron la remisión completa.
ConclusionesSe recomienda realizar la detección precoz de las infecciones por EBV y los NHL en todos los pacientes con diagnóstico de IDVC, independientemente de la edad del diagnóstico.
Immunodeficiencies are a very broad group of diseases with different pathogenetic mechanisms, which have in common a failure in the regulation of the immune defense against foreign aggressions. This failure in the regulation gives rise to diseases whose origin could be in the alteration of a receptor (for example, TOLL type), in the absence of an interleukin (IL), in the lack of function of the B lymphocytes (BL) or T lymphocytes (TL) or in the lack of a protein such as MBL (mannose-binding lectin). In the end, whatever the inductor genetic failure, the predominant symptomatology is the presence of infections, although these patients also have cancers of lymphoid origin and autoimmune diseases.1,2 Immunodeficiencies are classified into two categories: i) congenital or primary, almost always due to mutations or deletions of genes that tend to be hereditary but can also be de novo mutations; and (ii) acquired or secondary due to a disease that facilitates the loss of antibodies or lymphocytes. Primary immunodeficiencies are associated with the genetic deficiencies mentioned above. Examples of diseases that promote secondary deficiencies are persistent infections such as malaria, chronic diarrhea or nephrotic syndrome.3 Currently there have been > 70 clinical forms of primary immunodeficiencies identified. Due to recent advances in molecular biology, the molecular defects responsible for an important part of the primary immunodeficiencies are known, with undeniable advantages in terms of the possibility of an accurate diagnosis, prenatal diagnosis in affected families, genetic counseling and, in some cases, the possibility of treatment through gene therapy (Table 1).4,5
Principal characteristics of primary immunodeficiencies.
| Form of PID | Examples of diseases | Age at initiation | Infectious complications | Other manifestations |
|---|---|---|---|---|
| Predominant antibody immunodeficiency (humoral) |
| > 6 months Can debut in adolescence or during adulthood |
|
|
| Combined Immunodeficiencies (humoral and cellular) |
| < 6 months of life |
|
|
| Defects of phagocytes |
| Newborn or infancy |
|
|
| Complement defect |
| Any age |
|
|
PID, primary immunodeficiency; ID, immunodeficiency; AR, autosomal recessive; Ig, immunoglobulin; SCID, severe combined immunodeficiency; EBV, Epstein-Barr virus; CMV, cytomegalovirus; VZV, varicela-zoster virus.
The relationship between immunity and cancer is well known. Tumor cell antigens induce effector immune responses that destroy tumor cells. An immunological approach is increasingly used for the diagnosis and treatment of cancer.
The immune defense against cancer is initially mediated by innate immunity, composed of natural killer (NK) cells capable of destroying tumor cells. This function potentiates with interleukins 2 and 12 (IL-2, IL-12) and interferons. Macrophages eliminate tumor cells using the same mechanisms that eliminate bacteria. Activated macrophages synthesize tumor necrosis factor (TNF) that produces thrombosis of the tumor blood vessels. Antitumor adaptive immunity is fundamentally mediated by LT CD8 +, which are cytotoxic lymphocytes sensitized against tumor antigens capable of destroying cancer cells. This specific cytotoxic effector function starts with antigen-presenting cells (dendritic cells) and is potentiated by stimuli of the LT CD4+. Adaptive humoral immunity consists of specific IgG antibodies against tumor antigens, but usually has no effector function against the tumor and only serves for the diagnosis and control of tumor evolution.6
1.2Epstein-Barr virus (EBV) and lymphomasEBV is a double-stranded, ubiquitous DNA virus belonging to the herpesvirus family, which selectively infects BL, persisting in a latent state. EBV antigens are expressed differently, depending on the stage of differentiation of the BL and have different activity. These viral antigens are expressed in the nucleus, cytoplasm and surface of infected cells. The first antigens found in a latent infection are the Epstein-Barr nuclear antigens (EBNAs) of which EBNA-LP, EBNA-2, EBNA-3A,−3B and −3C are known and whose function is the stimulus of BL proliferation. Latent membrane proteins −1 and −2 (LMP-1 and LMP-2) have oncogenic properties because they facilitate genomic instability and promote the immortalization and transformation of the infected BL. EBV is a BL polyclonal activator, independent of the TL cooperating signals or dendritic cells. The in vitro BL infection promotes the formation of lymphoblastoid cell lines (LCL) immortalized due to the expression of the EBNA, LMP proteins and various non-coding miRNAs. LMP1 and LMP2A act as constitutively active receptors that imitate the CD40 signals of differentiation and of the BL antigen receptor, respectively. By this mechanism, EBV appropriates the physiological pathway of activation of the BL and promotes its proliferation and differentiation to memory cells in which the virus may persist for the lifetime of the infected individual.7
When a poor cytotoxic TL CD8 + response occurs due to an immunodeficiency of any type, an uncontrolled proliferation of infected BL takes place that increases the probability of errors occurring during DNA replication, including some that affect the oncogenes or tumor suppressor genes. The loci of the immunoglobulins are the most susceptible to suffer translocations. Translocation of the MYC gene to the locus of the immunoglobulins gives rise to an abnormal MYC expression associated with the malignant transformation and uncontrolled proliferation of neoplastic BL clones, which will constitute B-cell lymphoma. However, it is known that in patients with immune deficiencies without MYC translocations the viral proteins are sufficient to stimulate the production of BL clones that make up the B lymphomas.8,9
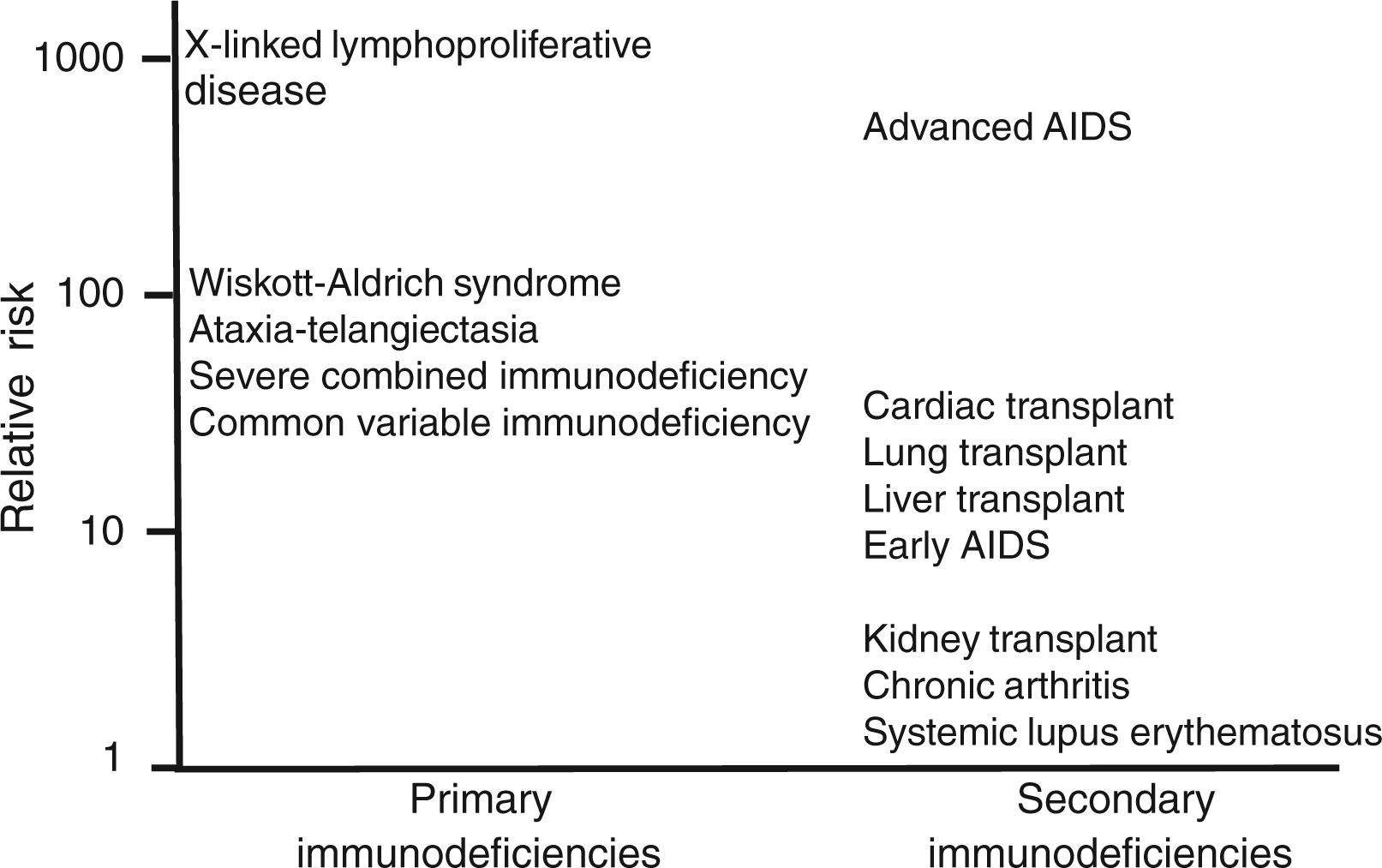
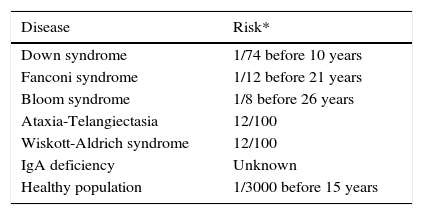
In situations of immune competence, tumor cells are eliminated by the defense mechanisms described and the virus persists asymptomatically, turning off its expression. Congenital and secondary or acquired immunodeficiencies are important risk factors for developing neoplasms. In Figure 1 and Table 2 the risk of leukemias and lymphomas in diseases accompanied by immunodeficiency are described.

Congenital diseases associated with an increased risk of leukemia or lymphoma.
| Disease | Risk* |
|---|---|
| Down syndrome | 1/74 before 10 years |
| Fanconi syndrome | 1/12 before 21 years |
| Bloom syndrome | 1/8 before 26 years |
| Ataxia-Telangiectasia | 12/100 |
| Wiskott-Aldrich syndrome | 12/100 |
| IgA deficiency | Unknown |
| Healthy population | 1/3000 before 15 years |
In the Hospital Universitario Sant Joan de Déu (HUSJD) the highest frequency of neoplasms in children with primary immunodeficiency has been detected in patients with common variable immunodeficiency (CVID) and in all cases has been associated with B cell lymphomas.10,11
CVID is a primary immunodeficiency characterized by an alteration in the differentiation of B cells, with a decrease in the production of immunoglobulins. It is the most prevalent form of severe antibody defects that presents itself at a rate of 1/25,000 to 1/60,000 individuals.12 CVID encompasses a heterogeneous mixture of disorders due to immunoglobulin and Toll-like receptors (TLR9 and TLR7) deficits that translate in lack of stimulation of dendritic cells and interferon alpha production, fundamental mediators in the initiation of the antitumoral innate immunological defense. Clinical manifestations are also heterogeneous and include recurrent infections (in almost 100% of patients), chronic lung disease (30–50%), autoimmune diseases (25%), gastrointestinal diseases (20%), allergic disease (33%) and elevated susceptibility to develop lymphomas. Disease initiation is typically described after puberty and before the age of 30 years, although there is a bimodal distribution with a peak between 1 and 5 years and another between 18 and 25 years. Diagnosis of the disease is established by clinical and analytical criteria and after making a differential diagnosis with other diseases presenting with hypogammaglobulinemia.13 Since 2003, mutations in four genes associated with CVID have been described, which would represent 10–15% of the cases. The mutations described in three of the genes present an autosomal recessive inheritance pattern and are extremely rare: deficit in the inducible co-stimulator of activated T-cells (ICOS), CD19 deficit and deficit in the B-cell activating factor (BAFF-R). The fourth group corresponds to mutations in the TNFRSF12B gene (TACI) described in up to 10% of patients with CVID, with an autosomal dominant, autosomal recessive or complex inheritance pattern (healthy relatives who are carriers of the mutation have been found).14
Because it is a very heterogeneous disease its prognosis is very variable. There are patients in whom infections predominate and are easier to treat and respond well to replacement therapy with subcutaneous or intravenous gammaglobulin. Another group of patients have more frequent autoimmune diseases. In the experience of the HUSJD, the most common autoimmune diseases in patients with CVID are type 1 diabetes mellitus, thyroiditis, celiac disease, systemic lupus erythematosus and vitiligo; sometimes several of these diseases coexist in the same patient. In these children, treatment with gammaglobulin does not adequately control the autoimmune disease that requires immunosuppressant or substitutive treatment of the disease itself. Children with neoplasms (lymphomas) have a prognosis derived from the optimal therapeutic response to the anti-CD20 (rituximab) monoclonal antibodies. Cytostatic treatment is necessary in cases in which there is no response to rituximab.
Regarding the prognosis, it should be kept in mind, as a whole, that treatment with gammaglobulin therapy reduces the number of infections, hospitalizations and improves quality of life. However, it has not been shown to protect from the development of neoplasms or non-infectious complications, such as autoimmune diseases or granulomatous disease.
2Clinical casesIn the experience of the HUSJD in Barcelona, Spain the highest frequency of neoplasms in children with primary immunodeficiency has been detected in patients with CVID, and in all cases the neoplasms have been B cell lymphomas.11 The experience in regard to B cell lymphomas in children diagnosed with CVID in the HUSJD is described below. Between the years 2000 and 2013 there were 28 children detected with CVID, diagnosed according to the criteria of the European Society for Immunodeficiencies (ESID) and the Pan American Group of Immunodeficiencies (PAGID).15 Of the 28 children with CVID, four had B cell lymphomas, two of them associated with EBV.
2.1Case 1We present the case of a Caucasian male patient. At 2 months of age he had chickenpox without complications. After 12 months of age he had mumps, bronchitis, otitis media, and recurrent conjunctivitis. At 2 years of age he underwent tonsillectomy and adenoidectomy for persistent otitis. Multiple eye and ear cultures were done, which were positive for Haemophilus influenzae, Streptococcus pyogenes, Streptococcus pneumoniae, Pseudomonas aeruginosa and Staphylococcus aureus at 4 years of age. At this same age he had mononucleosis syndrome due to cytomegalovirus (CMV). At 5 years of age he was diagnosed with CVID. Treatment with intravenous immunoglobulins was begun. He continued to have recurrent respiratory tract infections despite having correct levels of IgG. He demonstrated a lag in weight/height.
At 8 years of age he developed persistent cervical and inguinal adenitis. Biopsy of the inguinal lymph node showed evidence of follicular hyperplasia and PCR was positive for EBV. At 12 years of age he presented a new episode of persistent cervical adenitis. Biopsy of the cervical adenopathy confirmed a marginal non-Hodgkin’s lymphoma (NHL) with PCR positive for EBV. NHL was immunophenotype positive for CD20 and CD79. The extension study was negative. He was classified as stage III due to the pathological characteristics of the lymph nodes on computed tomography (CT). Treatment with rituximab at a dose of 375mg/m2 was decided upon with a first induction cycle of four weekly doses, followed by a phase of consolidation with four doses at monthly intervals and finally the maintenance phase with two doses a month until 2 years of treatment were completed. Complete remission was obtained.
The patient has maintained normal or elevated IgM levels, ranging between 500 and 1700mg/dl up to 10 months after the initiation of rituximab. Hyper-IgM syndrome was ruled out due to the correct expression of CD40 and CD40L and absence of genetic mutations, and X-linked lymphoproliferative syndrome due to the absence of mutations in the SH2D1A and XIAP genes.
2.2Case 2We present the case of a female African-American patient who presented with chronic diarrhea of unknown origin and somatic developmetal delay from early infancy. At the age of 8 years she was transferred to Spain and at that age was diagnosed with CVID. Physical examination revealed splenomegaly, pallor, cervicolateral lymphadenopathy, bilateral lung rales, hypogammaglobulinemia and anemia. After immunological study that showed decrease in isohemagglutinins and IgG, IgM and IgA, she was diagnosed with CVID and treatment was begun with intravenous immunoglobulins. Minimal duodenal lesions and lymphocytic inflammatory infiltration were noted on intestinal biopsy. Pulmonary function showed a mixed obstructive and restrictive pattern without response to bronchodilators. Lung CT showed multiple bilateral parenchymal and subpleural nodules, hilar lymphadenopathy, and mediastinal and axillary lymphadenopathy. Lung biopsy confirmed low-grade lymphoma of marginal zone, PCR positive for EBV and classified as stage II. Extension study was negative. She was treated with rituximab as in Case 1, with complete remission.
She persisted with chronic diarrhea due to the presence of colon ulcers with PCR positive for CMV on biopsies. At 14 years of age she presented enterocolitis with poor response to different treatments that required total colectomy with ileostomy. Histological analysis showed indeterminate inflammatory disease. Currently, at the age of 16 years, she remains stable, with secondary osteoporosis and lung function with mixed pattern.
During the diagnosis of CVID, the percentage of B cells in peripheral blood was 11%, and 3 years after rituximab is from 0–2%. Bruton gammaglobulinemia was ruled out by negativity of mutations in the Bruton tyrosine kinase gene (BTK).
2.3Case 3We present the case of a Caucasian male with an intense bout of chickenpox at 18 months of age and recurrent bronchitis until 3 years of age. At 4 years of age he was diagnosed with abdominal Burkitt lymphoma with liver and central nervous system metastasis (brain and leptomeninges), classified as stage IV. Chemotherapy was administered (vincristine, cyclophosphamide and prednisone, methotrexate, ARA-C and intrathecal hydrocortisone) along with cranial and spinal radiotherapy. After the initiation of chemotherapy he developed herpes zoster. At 6 years of age he was diagnosed with CVID and was started on replacement therapy of intravenous immunoglobulins. The patient had low stature due to growth hormone deficiency, probably secondary to skull radiation. He had bronchial asthma from 7 years of age and allergic rhinoconjunctivitis caused by seasonal allergies to wild grasses. Currently, at 13 years of age he is clinically stable, without infections or relapses of lymphoma. He continues with low stature and respiratory allergy. There is no information with regard to the EBV status.
2.4Case 4We present a female Caucasian patient with tonsillitis, otitis and pneumonia between 3 and 4 years of age and adenoidectomy at 5 years of age. At 11 years of age she presented with an inguinal tumor diagnosed as stage II marginal low grade B-cell NHL. Immunohistochemical study of the lymph nodes showed weak expression of CD20, CD45 and CD43 and positive follicular expression for bcl-2 and negative for CD10 (ruling out follicular lymphoma). There was monotypical expression of kappa chains and interfollicular plasmacytoid differentiation, without expression of cyclin D1 (ruling out mantle lymphoma). With in situ hybridization there were no translocations detected (14;18) or (11;14). Anti-EBV IgG positive antibodies were found in plasma. CHOP chemotherapy was begun (cyclophosphamide, doxorubicin, vincristine and prednisone) every 21 days for 6 cycles. Complete remission was obtained.
At 13 years of age she developed autoimmune hemolytic anemia difficult to control with steroids and which required two cycles of rituximab (375mg/m2/week, 4 weeks), with space of 6 months between relapses. At age 15 years, a year after completing rituximab, she developed B lym-phopenia and persistent panhypogammaglobulinemia. The study for autoimmune lymphoproliferative disease was positive and she was diagnosed with CVID. Treatment was begun with intravenous immunoglobulins. Subsequently she presented with cervical and inguinal adenitis and after a positron emission tomography (PET) and inguinal and axillary biopsies were done, reactive lymphoid hyperplasia was reported. At 16 years of age she developed right submandibular lymphadenopathy, confirmed by biopsy to be MALT type marginal lymphoma. EBV was negative for in situ hybridization and latent membrane protein was also negative by immunohistochemistry. Treatment was administered with rituximab. At 18 years of age she was transferred to an adult treatment center due to a recurrence of the MALT lymphoma. Tables 3 and 4 summarize the clinical, laboratory characteristics, type of lymphoma and treatment given.
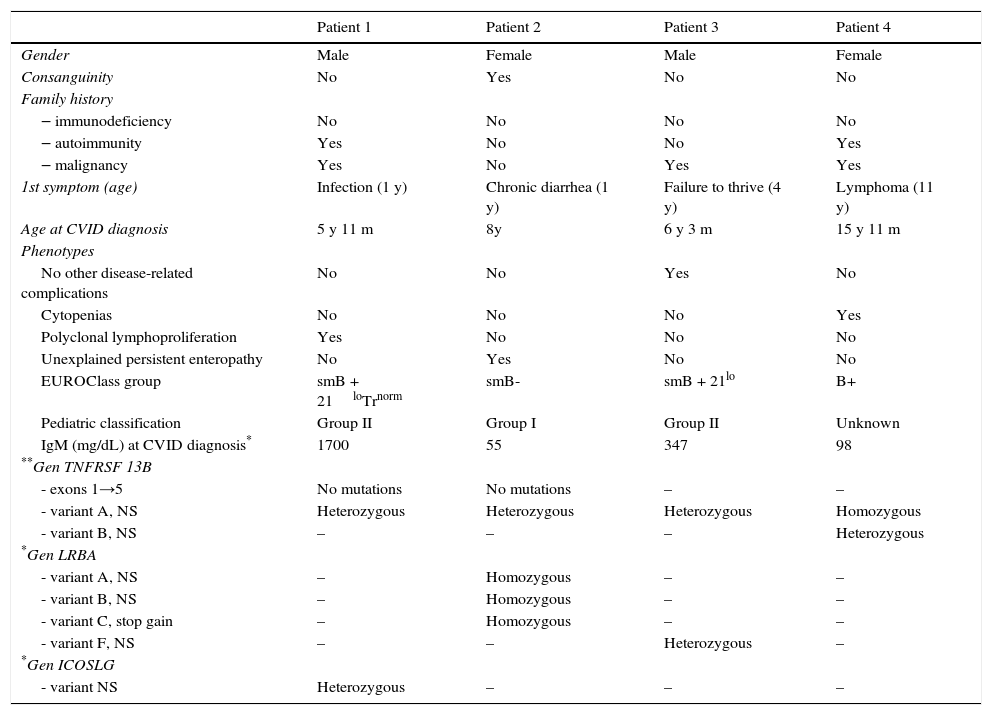
Clinical and laboratory characteristics associated with CVID patients who develop NHL.
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | |
|---|---|---|---|---|
| Gender | Male | Female | Male | Female |
| Consanguinity | No | Yes | No | No |
| Family history | ||||
| ‒ immunodeficiency | No | No | No | No |
| ‒ autoimmunity | Yes | No | No | Yes |
| ‒ malignancy | Yes | No | Yes | Yes |
| 1st symptom (age) | Infection (1 y) | Chronic diarrhea (1 y) | Failure to thrive (4 y) | Lymphoma (11 y) |
| Age at CVID diagnosis | 5 y 11 m | 8y | 6 y 3 m | 15 y 11 m |
| Phenotypes | ||||
| No other disease-related complications | No | No | Yes | No |
| Cytopenias | No | No | No | Yes |
| Polyclonal lymphoproliferation | Yes | No | No | No |
| Unexplained persistent enteropathy | No | Yes | No | No |
| EUROClass group | smB + 21loTrnorm | smB- | smB + 21lo | B+ |
| Pediatric classification | Group II | Group I | Group II | Unknown |
| IgM (mg/dL) at CVID diagnosis* | 1700 | 55 | 347 | 98 |
| **Gen TNFRSF 13B | ||||
| - exons 1→5 | No mutations | No mutations | – | – |
| - variant A, NS | Heterozygous | Heterozygous | Heterozygous | Homozygous |
| - variant B, NS | – | – | – | Heterozygous |
| *Gen LRBA | ||||
| - variant A, NS | – | Homozygous | – | – |
| - variant B, NS | – | Homozygous | – | – |
| - variant C, stop gain | – | Homozygous | – | – |
| - variant F, NS | – | – | Heterozygous | – |
| *Gen ICOSLG | ||||
| - variant NS | Heterozygous | – | – | – |
y, year; m, month; NS, nonsynonymous.
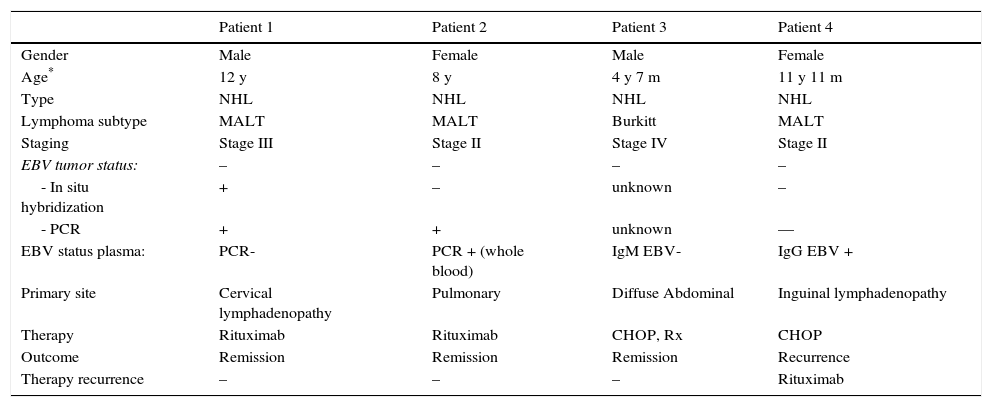
Lymphoma characteristics, treatment and outcome.
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | |
|---|---|---|---|---|
| Gender | Male | Female | Male | Female |
| Age* | 12 y | 8 y | 4 y 7 m | 11 y 11 m |
| Type | NHL | NHL | NHL | NHL |
| Lymphoma subtype | MALT | MALT | Burkitt | MALT |
| Staging | Stage III | Stage II | Stage IV | Stage II |
| EBV tumor status: | – | – | – | – |
| - In situ hybridization | + | – | unknown | – |
| - PCR | + | + | unknown | — |
| EBV status plasma: | PCR- | PCR + (whole blood) | IgM EBV- | IgG EBV + |
| Primary site | Cervical lymphadenopathy | Pulmonary | Diffuse Abdominal | Inguinal lymphadenopathy |
| Therapy | Rituximab | Rituximab | CHOP, Rx | CHOP |
| Outcome | Remission | Remission | Remission | Recurrence |
| Therapy recurrence | – | – | – | Rituximab |
y, years; m, month; NHL, non-Hodgkin lymphoma; MALT, mucosa-associated lymphoma tissue; EBV, Epstein-Barr virus; CHOP, cyclophosphamide, doxorubicin, vincristine, prednisone; Rx, radiation therapy.
Four children diagnosed with CVID who developed NHL are described. Some patients presented here have been previously described 11. Lymphoma was associated with EBV in two of the children. In addition, two patients developed lymphoma prior to diagnosis of CVID.
Complications of the CVID include an increased risk of malignancy, especially lymphoma and gastric cancer.16–19 Lymphoma can be considered one of the most serious complications of CVID. The cause of the increased risk of lymphoma in CVID seems to be multifactorial where the interaction of chronic infections, regulation of the immune system and genetic disorders facilitate the development of the malignant transformation.20
Kinlen et al.21 reported a 30-fold increase in the risk of developing lymphoma in CVID, whereas Cunningham-Rundles et al.17 reported an increase of 260-fold. Results of the data registry of Sweden and Denmark showed an increase of 12 times in the risk of lymphoma.22 The Italian study of Quinti et al. supports an increase of 18 times higher in the risk of lymphoma, with four cases of NHL during 5 years.12 The Australian study with 416 patients reported a standardized incidence rate (SIR) of 12.1 for each 1.132 of NHL patients (95% CI: 6.03–21.0).18
Lymphomas that occur in patients with CVID are most commonly of B-cell origin. There is a predominance of NHL, generally from the fourth to the seventh decade of life and rarely in children. It is commonly extranodal and EBV negative.23 A retrospective study of lymph node biopsies of patients with primary immunodeficiencies showed that 16 of 19 samples were positive for lymphoma and were patients diagnosed with CVID.24 The majority were NHL with only one case of Hodgkin’s disease and EBV was not found in any of the in situ samples by hybridization. Patients of this study were children < 18 years with the lymphoma diagnosed between 4 and 13 years of age (4 years of age for the Burkitt’s lymphoma and from 10 to 13 years for MALT lymphoma). The in situ hybridization for EBER1 of the EBV was positive in patient 1 and negative in patients 2 and 4. There is no information available on patient 3. EBV was found by PCR in the biopsies of patients 1 and 2.
The role of different members of the herpesvirus family has been investigated in patients with CVID. Wheat et al. reported an important finding: the presence of the human herpesvirus 8 (HHV-8) in six of the nine patients diagnosed with CVID with interstitial granulomatous lung disease and lymphoma (GLILD).25 One patient developed NHL and another developed MALT lymphoma. In the first patient the HHV-8 virus was found by qRT-PCR in a high number of copies in the malignant lymph nodes. Patient 2 of our cases had PCR study for pulmonary biopsy negative for HHV-8. Raeiszadeh et al. found an increase of CD8 + T cells specific for CMV peptides in patients with CVID compared with controls.26 Their observation was related with a higher prevalence of colitis and subsequent development of NHL. In our series, patient 1 was diagnosed with infectious mononucleosis by CMV before the CVID and the lymphoma. Patient 2 had PCR positive for CMV in colon biopsies years before lymphoma treatment.
Treatment of immunocompetent patients with nongastric MALT lymphoma usually includes immunotherapy with monoclonal antibodies directed to CD20 (rituximab) with or without chemotherapy. In patients with primary immunodeficiencies, standard chemotherapy regimens are not well tolerated. Anti-CD20 monoclonal antibodies could achieve complete remission in patients with B-cell lymphomas and CVID. Rituximab should be considered as monotherapy or associated with a reduced chemotherapy regime.27–29 Two of our patients were treated with rituximab as monotherapy achieving a complete remission and relapse-free after 3 and 6 years of follow-up, respectively. The third patient with MALT lymphoma was initially treated with CHOP. Treatment of the recurrence after 4 years of remission was done with rituximab, achieving a second complete remission.
In conclusion, CVID is a common congenital immunodeficiency with clinical manifestations of infections, autoimmune diseases and neoplasms. In pediatric patients it may initially present as a neoplastic disease, especially NHL, and is frequently associated with EBV infection. Early detection of the EBV infections and the NHL in all patients with diagnosis of CVID is recommended, regardless of the age at diagnosis.
Ethical disclosureProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of interestThe authors declare no conflict of interest.