



Introducción: La enfermedad de Pompe (EP) es un trastorno autosómico recesivo, causado por deficiencia de la enzima lisosomal, α-glucosidasa ácida (GAA) humana. Este déficit produce depósitos de glucógeno en hígado y músculos. La EP en niños pequeños (inicio temprano) produce cardiomiopatía e hipotonía. Sin embargo, en el inicio tardío, la cardiomiopatía se considera poco frecuente, y se ha dicho que hay menor beneficio con la terapia de reemplazo enzimático (TRE) con GAA.
Caso clínico: Paciente femenino de 8 años, que inició con síntomas a los 3 años de edad (inicio tardío): infecciones respiratorias recurrentes y debilidad muscular progresiva. Se diagnosticó con EP por evidencia en biopsia muscular y la baja actividad de α-glucosidasa. Se realizó lobectomía derecha basal por bronquiectasias y necrosis. Desarrolló neumonía y recibió asistencia respiratoria mecánica (CPAP) por 4 semanas, con dependencia absoluta de oxígeno y BPAP. Se aplicó TRE con GAA.
Conclusiones: La evolución positiva de la paciente, especialmente en las funciones respiratoria y cardiovascular, muestra que es posible mejorar la cardiomiopatía en pacientes con EP de inicio tardío.
Background: Pompe disease (PD) is an autosomal recessive disease caused by a deficiency in the lysosomal human enzyme α-alglucosidase. Among children (early onset), PD causes cardiomyopathy, whereas late-onset disease seems unrelated to a high rate of cardiomyopathy. Patients respond less to enzymatic replacement therapy (ERT) with α-alglucosidase.
Case report: This is the case of an 8-year-old female patient with symptom onset at 3 years of age (late onset) with recurrent respiratory infections and progressive muscular weakness. Diagnosis for Pompe disease (PD) was established due to evidence from muscle biopsy. At baseline, a right lobectomy was performed for bronchiectasis and necrosis. The patient developed pneumonia and received mechanical respiratory support (CPAP) for 4 weeks with absolute dependence on oxygen and BPAP. ERT with α-alglucosidase was given.
Conclusions: This patient's positive output and remarkable effects on cardiovascular and respiratory function suggest that ERT may reduce cardiomyopathy among late-onset PD patients.
Introduction
Lysosomal storage diseases are a group of inborn errors of metabolism that may be autosomal recessive or X-linked of genetic origin. The effect is an enzymatic deficit that allows abnormal accumulation of non-degraded substrate and causes damage and specific clinical manifestations in different organs. These effects are considered to be rare and exhibit very low incidence rates, but they are a public health problem due to causing premature death, have a chronic disease course, cause significant neurological disorders and deterioration of the quality of life.1,2
Until 20 years ago, only treatment for complications was provided, so its behavior was expectant and its course could not be changed. At present, with the emergence of enzyme replacement therapy (ERT), a door has been opened to the possibility of changing the course of this group of diseases.
The enzyme deficit makes ERT the main treatment resource for clinical management. An example of its efficacy was recorded in Gaucher disease (GD). It has only been two decades that the only existing therapy was palliative for management of complications. At present, ERT is today a milestone in the treatment of the GD for its ability to control symptoms, reduce disability and improve the quality of life.1,2
Pompe disease
PD was described by the Dutch pathologist Joannes C. Pompe. It is also known as "glycogenosis type II" (GSDII). In 1932, Pompe published the clinical case of a 7-month-old patient with cardiomegaly who died due to apparent complications of pneumonia and whose biopsy showed massive glycogen deposits mainly affecting skeletal and heart muscle. Three decades later (1963), Herz observed the vacuoles with glycogen deposit with electron microscopy and described the disease according to liposomal deposit. On the face of these findings, it can be said that the muscular condition is secondary to glycogen deposit in the liposomes because of a deficit in acid α-glucosidase (GAA).3
Clinical manifestations
The cardinal clinical manifestations of PD are weakness and hypotonia, for which reasons it is also considered to be a neuromuscular disease or metabolic myopathy. Its phenotypic variability has led to it being classified into two types according to the age of presentation: PD of early onset and late onset. At present, PD is considered as a continuous aspect of manifestations that vary according to the residual activity of GAA and which leads, invariably, to progressive weakness and death due to glycogen accumulation.3,4
On average, early-onset PD starts at 2 months of age. Diagnosis is made at 4 months and the time for the first ventilatory support is 5.9 months and with survival of 8.7 months. Without treatment, PD has an average mortality of 74% at 1 year of age and 88% at 18 months. Death is usually due to cardiac insufficiency with generalized hypotonia and respiratory muscle weakness. In the heart there is progressive thickening of the left ventricle (LV) due to deterioration of systolic function and development of progressive cardiac insufficiency that causes death before 1 year of age. Different authors consider early-onset PD as infantile PD and late-onset disease as juvenile PD.3,5,6
A study carried out in a series of 19 patients with infantile (early onset) PD showed a short PR interval in up to 75% of the cases as well as the fact that interference with conduction tissues promotes very high complex QRS in the electrocardiogram. These alterations in cardiac conduction and ventricular hypertrophy increase the risk of sudden tachyarrhythmias, which can be fatal in situations of increased cardiac output. These patients also showed a normal intellect.5,6
Enzyme replacement therapy in Pompe disease
ERT has been available since the year 2000 due to recombinant DNA technology. In 2007, a study with 18 patients <6 months of age with PD and cardiomyopathy treated with ERT based on GAA for 52 weeks showed a reduction in the use of assisted ventilation and in mortality. Later clinical studies showed that ERT substantially improves survival rates and function of respiratory muscles as well as reduces disability due to skeletal muscle weakness.5,7
In early-onset PD, ERT has demonstrated to influence myocardial remodeling, improve ventricular function and decrease wall thickness. In late-onset PD, cardiac involvement is more frequent for which reason the efficacy of ERT is estimated to be limited without reports in the literature of significant improvement in function and ventricular size.8
Due to the progress achieved with ERT in lysosomal storage diseases, it is important to determine whether there are significant changes in remodeling and myocardial function in patients with late-onset PD. This experience with ERT administration in a female Mexican patient with late-onset PD with a good response and remission of heart damage is, from this perspective, an exceptional case that should be reported in the national and international literature.9-11
On this basis, the objective of this study was to describe the evolution of myocardial function in a Mexican patient, evaluating the benefit of reversal of cardiomyopathy with ERT and comparing the results of myocardial function with late-onset ERT with the international literature reports. Also, we describe the evolution of the musculoskeletal function including the respiratory response and its evolution from the onset of ERT.
Clinical case
The case presented is that of a 9-year-old female patient originally from the State of Mexico who was admitted to the Hospital Infantil de Mexico Federico Gómez in 2011 with a history of healthy, nonconsanguineous parents. The patient was from a low socioeconomic status and rural environment. There was no remarkable perinatal history and with age-appropriate psychomotor development.
Personal medical history revealed recurrent respiratory tract infections diagnosed as asthma at 3 years of age. She had four severe episodes of pneumonia. Upon admission she presented with orthopnea of 4 years evolution, which had increased during the last year. The patient required two to three pillows to sleep and had dyspnea and cough of 4 years evolution. She also had recurrent falls during the previous 6 months evolution, recurrence of respiratory tract infections and progressive edema of the face and perioral cyanosis.
She was admitted to the emergency department with a clinical picture of pneumonia and pleural effusion, cardiac insufficiency, and marked muscular weakness. She walked with assistance. Laboratory examination of March 2011 reported CBC and serum electrolytes within normal limits. Liver function test showed values of lactic dehydrogenase 688, total bilirubin 0.75, alanine aminotransferase 217 and aspartate aminotransferase 511. Arterial blood gases showed pH 7.39, pCO2 46, pO2 58, saturation 89%, and HCO3 26.6.
Imaging tests
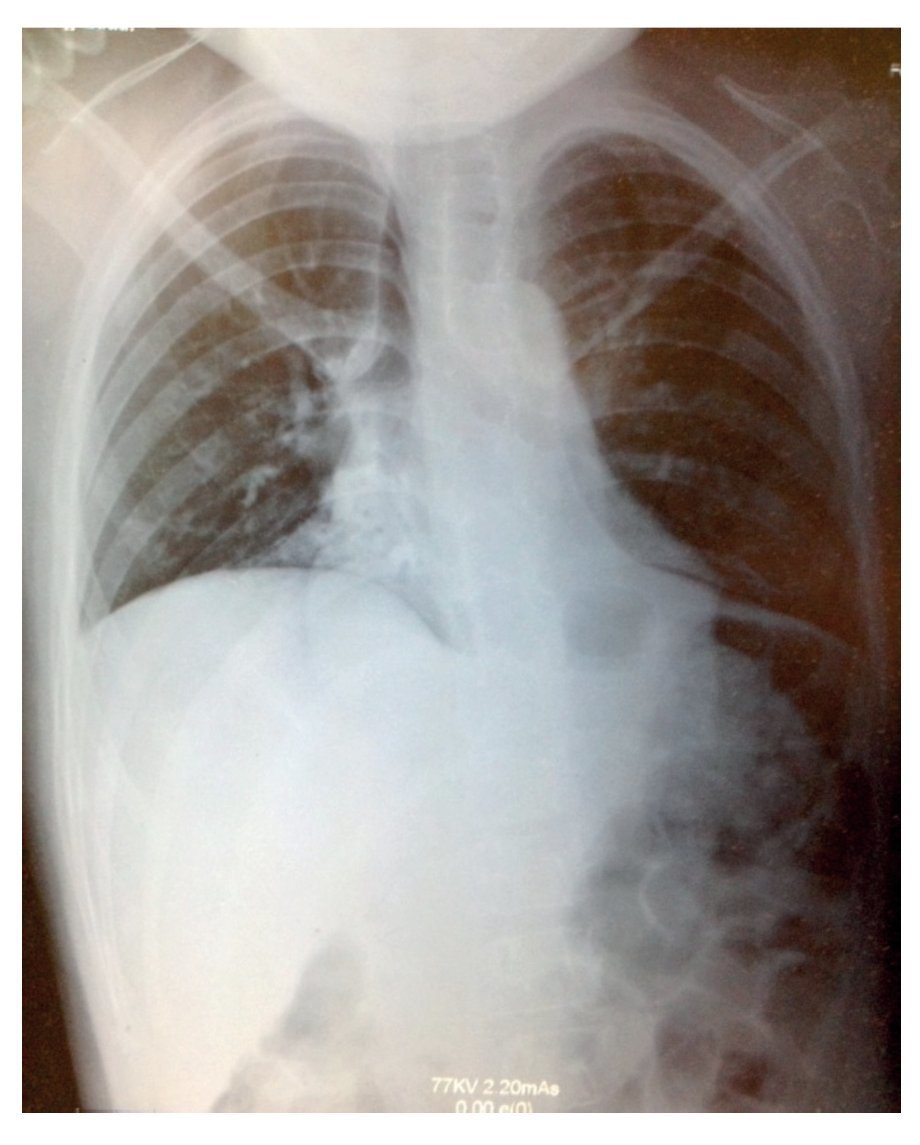
Plain x-ray showed data compatible with pneumonia: chest with right micronodular infiltrate with basal predominance (Fig. 1). Chest tomography with pulmonary window showed image suggestive of bilateral, basal and medial consolidation with air bronchogram as well as right pleural effusion without compromise of function (<10%).
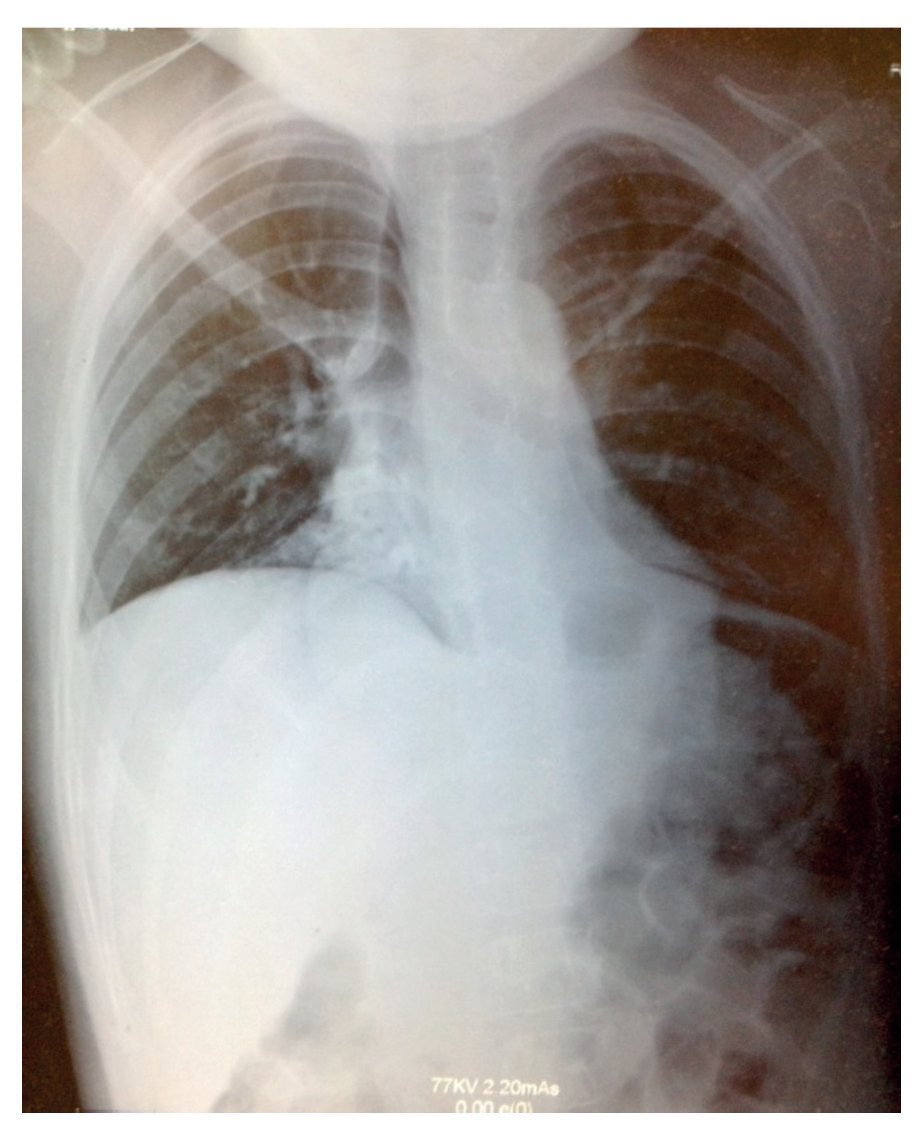
Figure 1 X-ray showing data compatible with pneumonia and chest with right micronodular infiltrate of basal predominance.
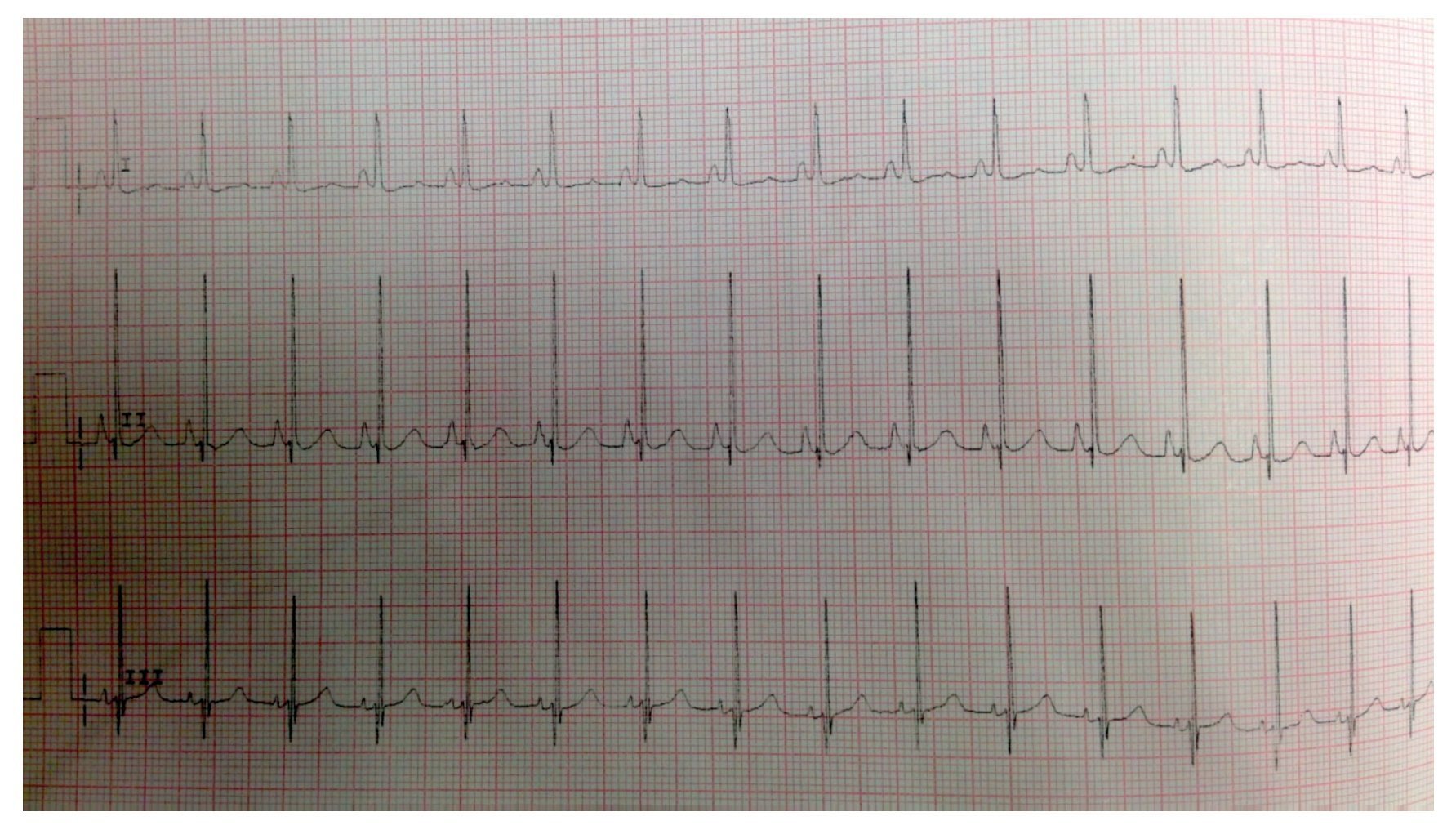
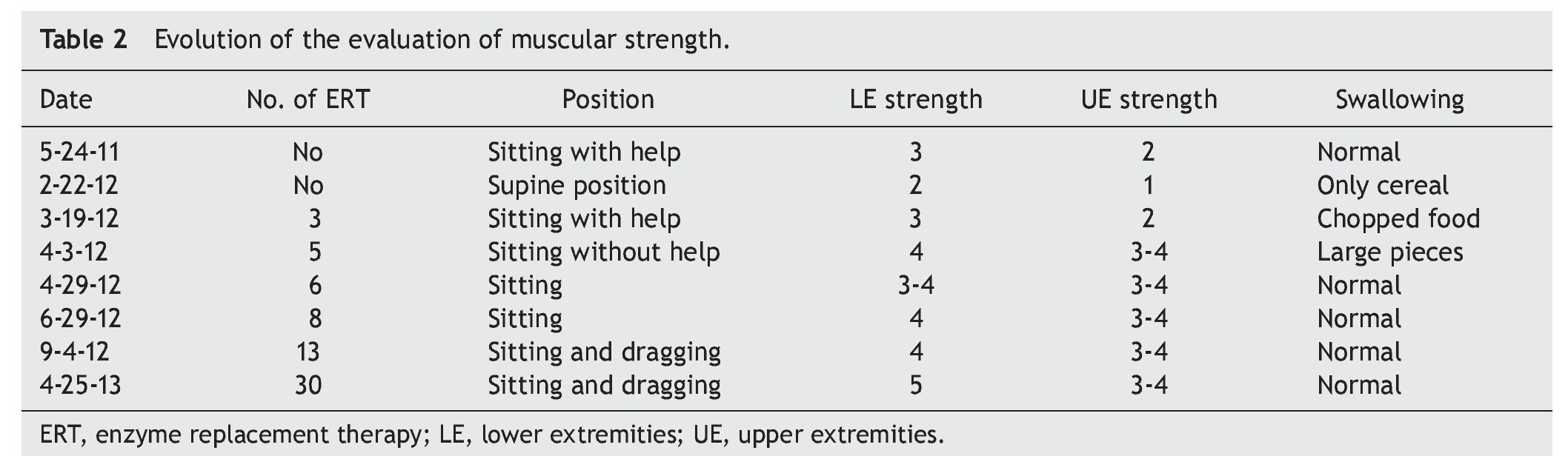
EKG showed sinus rhythm, biventricular hypertrophy, right atrial enlargement and ventricular repolarization disorder (Fig. 2). Transthoracic echocardiogram (TTE) was carried out that reported segment analysis normal for situs solitus without structural defects and normal ventricular function with ejection fraction (EF) of 78%. Right ventricular systolic pressure (RVSP) was estimated based on tricuspid valve regurgitation of 80 mmHg. Qualitative dilatation of right cavities and pulmonary artery trunk and branches was also observed.
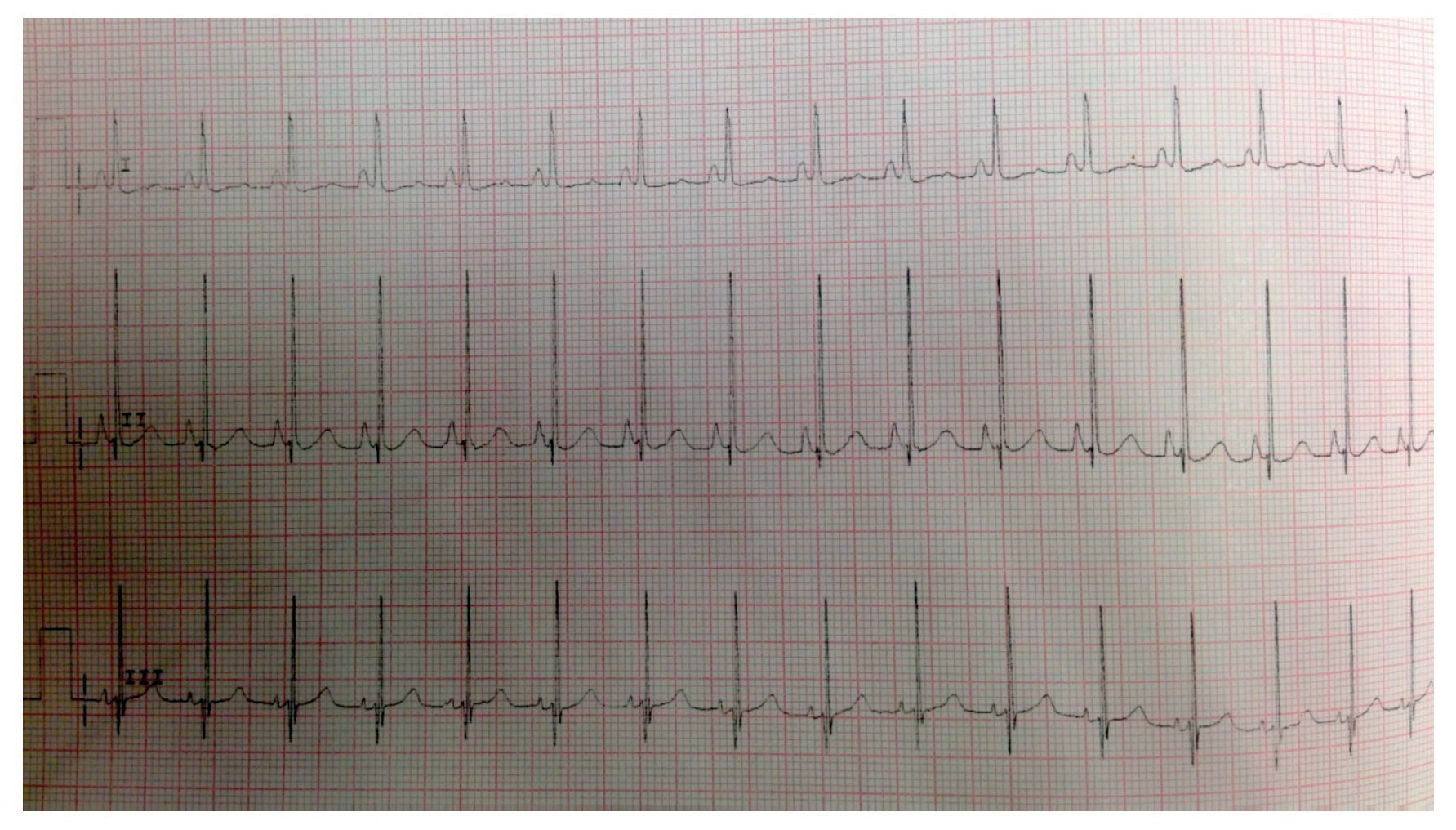
Figure 2 Electrocardiogram with flattened ST segment, Q DIII waves, aVF with T-wave inversion indicating right overload.
The findings of chronic pulmonary disease led to the clinical approach to rule out common causes such as primary immunodeficiency, cystic fibrosis, gastroesophageal reflux disease and mycobacterial infection. Some of the results are presented below:
• Spirometry: mild to moderate air entrapment, barely assessable using positive pressure, with important restrictions.
• Bronchoscopy and bronchioalveolar washing: hypopharynx, glottis, and arytenoid inclusion without alterations, with trachea and bronchi of normal appearance.
• Serial BAAR: negative.
• Sudan III: negative.
• Invasive forms in bronchoaspirate with blastoconidias and pseudomycelia without clinical correlation.
• ELISA study: negative.
• Immunoglobulins and complement: normal for age.
• Esophagogastroduodenal series: without alterations.
• Sweat electrolytes: Cl 11.6 mEq, Na 9.3 mEq, K 2 mEq, ruling out cystic fibrosis.
• Ventilatory perfusion scan: data associated with pulmonary hypertension and mixed pathology. Bibasal cylindrical bronchiectasis.
• Basal lobectomy: left lower lobe with scant fibrosis, macroscopic changes of the pulmonary parenchyma, superior lobe and lingula with normal characteristics.
Postoperatively, the patient presented alveolar hypoventilation and respiratory acidosis. Mechanical ventilatory support was required for 4 days, with 23 days stay in the intensive care unit. Patient had a convulsive crisis secondary to hypercapnea (pCO2 139). Ventilation was provided with BiPAP (bilevel positive pressure system), ruling out cystic fibrosis. In flow cytometry tests for lymphocytes, common antibodies from an autoimmune disease, complement, ANA, anti-DNA, pANCA and cANCA were observed. Complement and flow cytometry were reported to be within normal limits for age.
Before the progressive muscle weakness, serology for Trichinella spiralis was indicated, which was positive; however, the finding was ruled out when it did not correlate with cardiac problems and the result of the CPK. The approach for myopathy was continued. Electromyographic study for motor and sensory neuroconduction of the four extremities was within normal parameters, but the muscular fiber study was abnormal, showing typical inflammatory changes. Polysomnography was done due to the suspicion of obstructive apnea, which was confirmed due to CO2 retention and hypoxia.
When muscular biopsy was done (quadriceps), massive cytoplasmic inclusions of glycogen were found. It was concluded that it was a type II glycogenesis or PD. Diagnosis was confirmed with the determination of enzymatic activity. Values obtained were the following:
•α-glucosidase with pH 3.8: 0.24 (reference values 1.5-10 nmol/spot of 21 h);
•α-glucosidase with pH 7.0: 1.59 (reference values of 1.8-17.1 nmol/spot of 21 h);
•α-glucosidase with inhibition: 0.03 (reference values 0.9-7.2 nmol/spot of 21 h).
DNA sequencing results confirmed two mutations described for PD: C.258dupC (heterozygous) and C.1445C>T (heterozygous). Before initiating ERT, the patient had two episodes of pneumonia and cardiorespiratory arrest. She required advanced resuscitation, intubation and mechanical ventilation for 4 weeks. After various failed attempts, extubation was achieved and was continued with support in BiPAP mode during the day and during sleep, alternating with oxygen by nasal cannula. She received parenteral nutrition and gastrostomy because of inability to swallow due to muscular weakness and compromised nutritional state. On February 20, 2012, ERT was decided upon with an infusion of α-alglucosidase (20 mg/k/every 2 weeks).
Evolution of enzyme replacement therapy during the first year of treatment
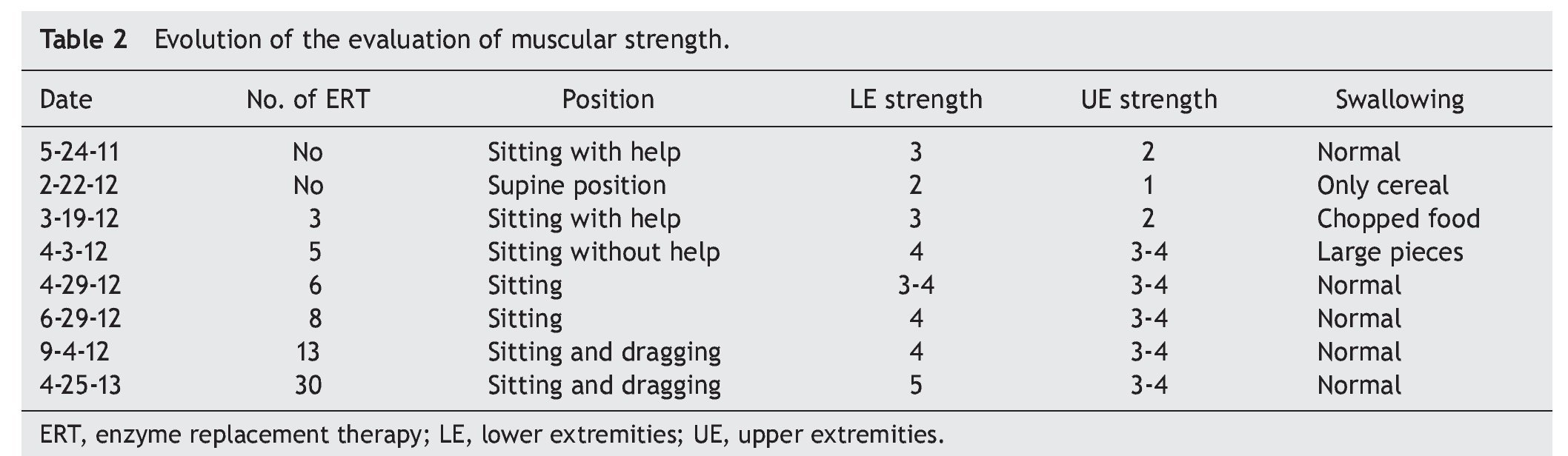
At the beginning of ERT, evaluation by rehabilitation showed a severe musculoskeletal deficiency limiting daily activities and social participation. Complete passive mobility arches were appreciated with strength 2/5 in most muscles of the trunk and upper extremities and 1/5 in the lower extremities according to the Daniels scale (Table 1).

One week after the first enzymatic infusion there was recorded progress of ventilatory mode, from SIMV to CPAP as tolerated. Swallowing also improved, progressing to a normal bland diet that was partially tolerated. Towards the end of the first month there was improvement in muscular strength recorded although respiratory effort was insufficient to remove assisted ventilation. Towards the sixth week, ventilation was changed to a BiPAP modality.
At 2 months there was a notable improvement recorded with regard to trunk support, sitting and mobility of the lower limbs according to the Daniels scale, with intrinsic muscle strength and prone supinators of 3; triceps, biceps, flexors and shoulder abductors of 2, hip flexors of 3, and knee extensors, eversion, flexors and extensors of the ankle of 1 (Table 2).
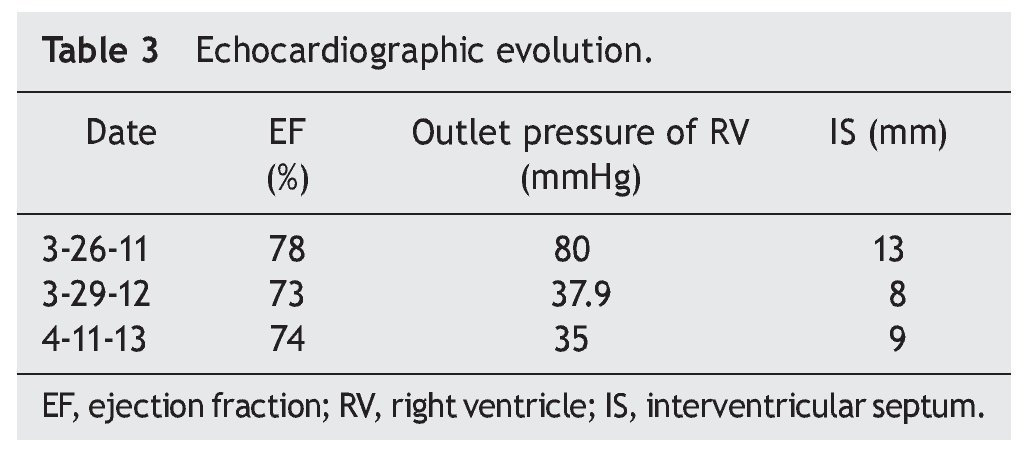
Towards the third month of ERT the BiPAP was able to be intermittently stopped with nasal cannula oxygen. The patient was discharged to continue with outpatient management, with treatment for pulmonary hypertension with anticongestives and pulmonary vasodilator as well as respiratory management with intermittent BiPAP. The patient had balance of the neck and trunk as well as support of hands. She progressed from a lying position to sitting with difficulty, moving short distances by crawling, and initiating independent feeding. The upper and lower extremities were still limited to knee extension with a knee rotation of 30º. Muscular strength was evaluated with the Daniels scale (Table 1) in the upper extremities (biceps, pronosupinators, and hand in 3, remainder 2, strength in lower extremities 2).
Towards the fifth month, mobility improved with crawling and dragging. An echocardiogram reported frank improvement in thickness of the septum and ventricular wall. When the semester of ERT was completed, there was a clear improvement in posture of the back, better respiratory effort and absence of fatigue. Two hours of inhaled oxygen were interspersed with 2 h of BiPAP. The patient showed better muscular tone in the legs. Three hours of BiPAP were alternated with 3 h of nasal cannula oxygen. The patient slept with the BiPAP. In light of the evidence of improvement of pulmonary hypertension it was decided to suspend treatment with sildenafil and furosemide, closely monitoring for any data of relapse.
At the end of the first semester with ERT, the patient presented clinical data of precocious puberty (hirsutism, breast button and axillary hair). The hormone profile was assessed and reported as normal.
Eight months later the patient used only BiPAP during sleep and nasal cannula oxygen was stopped. Arterial blood gases reported pH 7.38, CO2 36, O2 144. An improvement was noted in regard to ventilatory status with decrease of CO2 retention.
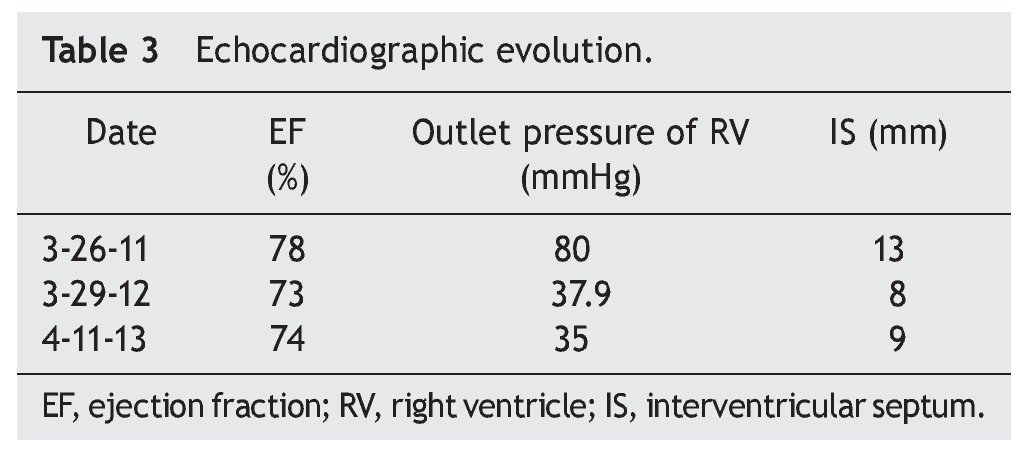
When the first year of ERT was completed, an echocardiogram was done and showed decreased thickness of interventricular septum (8 mm), RVSP 35 mmHg and systolic function of the same, RV preserved, with EF of 74% and reduction fraction of 42% (Table 3). Currently the patient is undergoing rehabilitation and improvement in motor function is evident. She also benefits from attending school. In general, the patient's quality of life has improved considerably.
Discussion
PD demonstrates a wide clinical spectrum from glycogen deposition in muscle and visceral tissue. It is evident that cardiac function in the early-onset variant has an extreme compromise, but satisfactory results can be obtained with ERT, achieving myocardium remodelling and ventricular function as well as decrease in the thickness of the ventricular wall. In late PD, generally, cardiac involvement is described as infrequent or less severe, and the results of ERT with regard to cardiac function have not been encouraging. Improvement in function or ventricular size has not been described. In this clinical case there is obvious involvement of the heart muscle as well as heart failure at disease onset (as shown in the echocardiogram) with right ventricular hypertrophy and pulmonary hypertension, documenting in addition outlet pressure of the RV of up to 80 mmHg. In contrast to existing literature reports where a significant change is not reported in left ventricular function or wall thickness between patients with late-onset PD, this patient showed that ERT can achieve a significant reduction of the RV output pressure from 80 mmHg to 35 mmHg. Also, a decrease in initial thickness of the interventricular septum is reported from 13 mm to 9 mm. With the reversal of heart failure, anticongestive and vasodilator treatments were suspended.
PD is a disease that, due to its low incidence and its wide spectrum of clinical presentation, is a diagnostic challenge for the pediatrician. Importance of an early diagnosis is associated with the timely start of specific treatment to avoid progression of the disease towards a disability of the skeletal, respiratory, and cardiac muscles and death as well as to promote a better quality of life. Due to the few cases reported and relatively recent discovery of ERT, its range of action is not yet completely known. As shown in this clinical case, it appears possible to achieve cardiac remodeling with improvement in ventricular function, even in patients with late-onset PD. Additional clinical evidence of progression of cardiomyopathy with the use of ERT in patients with late-onset PD is needed. It is possible to integrate patients with PD back to a normal life with ERT-based treatment. The general status of the patient improved. Gradually, the patient recovered 13 kg and 12 cm in 18 months of ERT.
Her physical activity increased, she attends school and her quality of life has significantly improved.
Conflicts of interest
The authors have no conflicts of interest to declare.
Received 22 October 2013;
accepted 12 December 2013
* Corresponding author.
E-mail:magyceron@yahoo.com (M. Cerón-Rodríguez).