





A 7-month-old female patient with failure to thrive and nonspecific metabolic disorders was seen in the gastroenterology clinic of the Hospital Infantil de México Federico Gómez (HIMFG).
1.1HistoryHer mother was 21 and her father 24 years old. She had a 3-year-old brother; they had not known disease neither evidence of consanguinity. She was breastfed for a month and complementary feeding since 5 months of age.
DEVELOPMENT. She had incomplete head control, did not roll over and did not sit. She attended rehabilitation therapy.
INMUNIZATIONS. BCG, a dose of acelullar pentavalent vaccine (DPaT+VIP+Hib) and two doses of hepatitis B vaccines.
She was the product of the second pregnancy with prenatal care since the first month of pregnancy. The mother received supplementary iron and folic acid. She was born in a hospital at 40 weeks of gestation, weight 3,050g, height 49cm, and an Apgar score of 8/9. She was discharged in the second day of life without the mother, who remained hospitalized five days because of hypovolemic shock.
At 2 months of age, she was hospitalized for 15 days because of an acute gastroenteritis with dehydration; she received antibiotics and a red blood cell transfusion because of anemia. Three months later, she was hospitalized again because of an increase of the abdominal circumference. An abdominal ultrasonography detected hepatomegaly, so she was sent to HIMFG. The referral note registered a weight of 4,800g, a height of 57cm, a head circumference of 41cm and the liver edge at 4-3-4cm below costal margin.
On admission to HIMFG the abdominal circumference had increased from 46 to 50cm in 8 days, and abdominal wall collateral vessels were visible. She had jaundice noticeable in mucous membranes and sclera which started 4 days prior to her arrival.
In the next two days, she showed progressive neurological deterioration: hypoactivity, drowsiness and stupor, increase in sleeping periods without response to awakening efforts. The following data were recorded: weight 4,000g, height 61cm, heart rate 153/min, respiratory rate 30/min, blood pressure 80/50mmHg, temperature 37.7°C, abdominal circumference 53cm, Glasgow coma score (GCS) of 9.
Her apparent age was under her chronological age. She was drowsy, with a normal head, normal pupils and icteric sclera. She had no lymphadenopathies, respiratory movements were asymmetric. Lungs were well ventilated. Heart sounds were increased in frequency with no murmurs. The abdomen was distended due to ascites; it was soft, compressible, and painless, and peristalsis could be heard. Extremities were normal, with good capillary refill, and adequate pulse intensity. Painful stimulation was needed for eye opening, verbal and motor response.
Ophthalmological exploration showed bilateral nuclear cataract.
The patient remained in critical care for 2 months. On admission, an endotracheal tube was placed, and although it could be removed, the patient developed ventilatory impairment associated with the restrictive ascites, abnormal fluid distribution and nosocomial infectious events. The ventilatory strategy was dynamic and included high frequency ventilation at the end of her stay.
She presented several events of hemodynamic instability, some related to nosocomial infection. She required management with vasopressors and inotropes. Ampicillin resistant Enterococcus faecium was identified in a blood culture. She received several broad-spectrum antibiotics at meningeal doses because of the impossibility to perform a lumbar puncture to rule out central nervous system infection. Given her risk factors and the presence of hyphae in urine, antifungal coverage was also included.
From a metabolic point of view, the patient coursed with hyponatremia of difficult management and persistent hypoglycemia. On the hematologic, she had anemia with anisocytosis, acanthocytosis and macrocytosis in peripheral blood smear. She had bleeding form mucous membranes associated with thrombocytopenia, liver failure coagulopathy and disseminated intravascular coagulation, which was managed with vitamin K, plasma and cryoprecipitate transfusions (Table 1).
General laboratory tests.
| Hb | Hct | Leu | Neu | Immature Neu | Lymph | Mon | Alfa 1 antitrypsin | PT | PTT | INR | LDH | Colesterol | Triglycerides | HDL | LDL | BUN | Creat | DB | IB | TB | ALT | AST | DHL | Alb | T Prot | BD | BI | BT | ALT | AST | Alb | Prot T | Gluc | Ammon | GGT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7.10g/dl | 22.8% | 37,300 | 42% | 8% | 24% | 26% | 106mg/dl | 31.6” | >120” | 2.43 | 477U | 67mg/dl | 38mg/dl | 33mg/dl | 26.4mg/dl | 19mg/dl | 0.5mg/dl | 10.6mg/dl | 12.7mg/dl | 23.3mg/dl | 27U | 40U | 524U | 4g/dl | 5.2g/dl | 9.9mg/dl | 12.7mg/dl | 22.6mg/dl | 31U | 75U | 3g/dl | 8.3g/dl | 61mg/dl | 144–259μmol/l | 27U |
Hb, hemoglobin; Hct, hematocrit; Leu, leucocytes; Neu, neutrophils; Lymph, limphocytes; Mon, monocytes; PT, protrombin time; PTT, partial tromboplastin time; LDH, lactic dehydrogenase; HDL, high density lipoproteins; LDL, low density lipoprotein; BUN, blood urea nitrogen; Creat, creatinin; DB, direct bilirubin; IB, indirect bilirubin; TB, total bilirubin; ALT, alanine aminotransferase; AST, aspartate aminotransferase; Alb, albumin; TProt, total protein; Gluc, glucose; Ammon, ammonia; GGT, gammaglutamyl transferase.
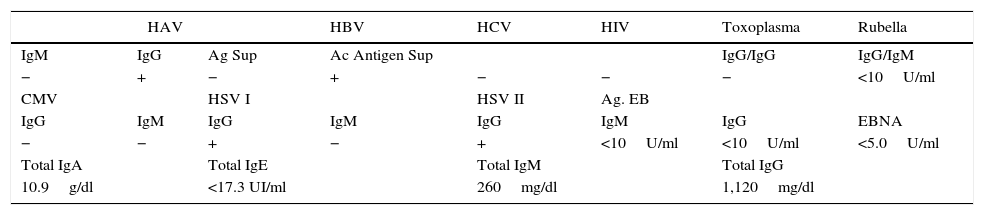
Within the approach for hepatopathy, portal hypertension without splenomegaly was identified. She continued with increasing abdominal distention, elevated aminotransferases and ammonium, hypoglycemia, with no acholic stools neither choluria (Table 1). Structural and infectious causes of liver disease were ruled out (Table 2). Amino acid and acylcarnitine profiles were reported as normal. Thyrotorpin (TSH) was 0.9mUI/ml and 25-OH vitamin D, 27.7ng/ml.
Serologic tests.
| HAV | HBV | HCV | HIV | Toxoplasma | Rubella | ||
|---|---|---|---|---|---|---|---|
| IgM | IgG | Ag Sup | Ac Antigen Sup | IgG/IgG | IgG/IgM | ||
| − | + | − | + | − | − | − | <10U/ml |
| CMV | HSV I | HSV II | Ag. EB | ||||
| IgG | IgM | IgG | IgM | IgG | IgM | IgG | EBNA |
| − | − | + | − | + | <10U/ml | <10U/ml | <5.0U/ml |
| Total IgA | Total IgE | Total IgM | Total IgG | ||||
| 10.9g/dl | <17.3 UI/ml | 260mg/dl | 1,120mg/dl | ||||
HAV, hepatitis A virus; HBV, hepatitis B virus; HCV, hepatitis C virus; HIV, human immunodeficiency virus; CMV, cytomegalovirus; HSV I, herpes simplex virus I; HSV II, herpes simplex virus II; EB, Epstein-Barr.
GCS was 12 while not on sedatives. Management included sodium restriction to 2mEq/kg/d, spironolactone 6mg/kg/d, furosemide 1mg/kg/d, lactulose 1ml/kg/d, rifaximin 20mg/kg/d, vitamin K 10mg/d, plasma and cryoprecipitate transfusion and protein restriction. Albumin infusion at 1g/kg was administered for ascites to manage the restrictive ventilatory pattern.
Renal function deteriorated: serum creatinine increased to 0.7mg/dl (Table 3), while fractional excretion of sodium (FeNa) was 0.6% and urinary sodium 6mmol/l. Hepatorrenal syndrome was diagnosed and was managed with terlipressin and albumin infusion. The abdominal pressure was 13cmH2O, and a therapeutic paracentesis was performed with 1,200ml drainage, which improved renal function.
Renal function tests.
| BUN | Creat | Na | K | Cl | Ca | P | Mg | |
|---|---|---|---|---|---|---|---|---|
| Blood | 7.5mg/dl | 0.5mg/dl | 146mEq/l | 4.1mEq/l | 101mEq/l | 8.3mg/dl | 3.7mg/dl | 1.9mg/dl |
| Urine | 204mg/dl | 10.2mg/dl | 50mEq/l | 20.9mEq/l | 54mEq/l | 9.3mg/dl | 36.3mg/dl | |
| Urianalysis | Cetone + | Glucose + | ||||||
BUN, blood urea nitrogen; Creat, creatinine; Na, sodium; K, potassium; Cl, chloride; Ca, calcium; P, phosphorus; Mg, magnesium.
The algology service prescribed morphine 100mg/kg/dose. After 2 months of in-hospital stay with intensive management, she had a cardiorespiratory arrest. No resuscitation attempts were performed at the request of the parents.
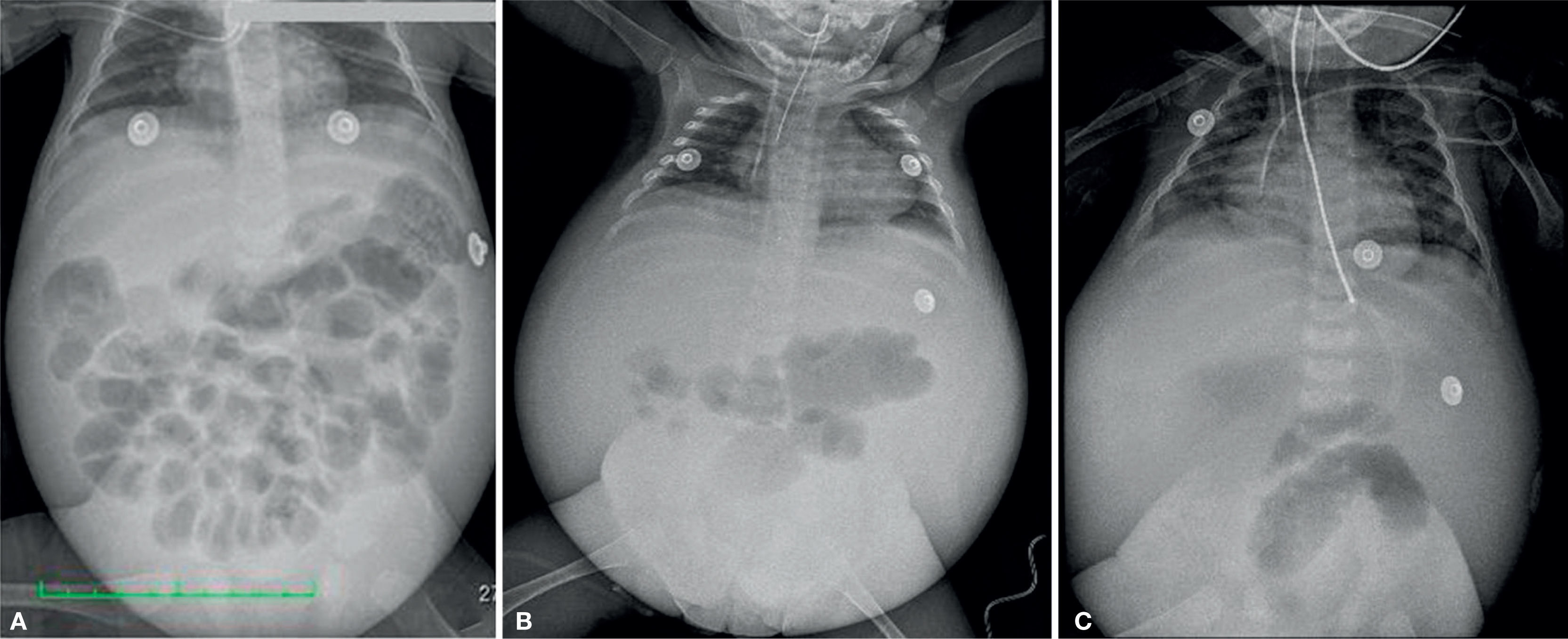
2ImagingCephalic displacement of bowel loops can be observed in thoracoabdominal radiographs, along with opacity of the pelvic cavity, intestinal dilated loops and decreased lung volume because of diaphragm elevation. Radiopacity of the abdomen increased over time in relation to the increase of the ascitic fluid. Ribs are horizontalized and the heart shadow is enlarged. (Figure 1).

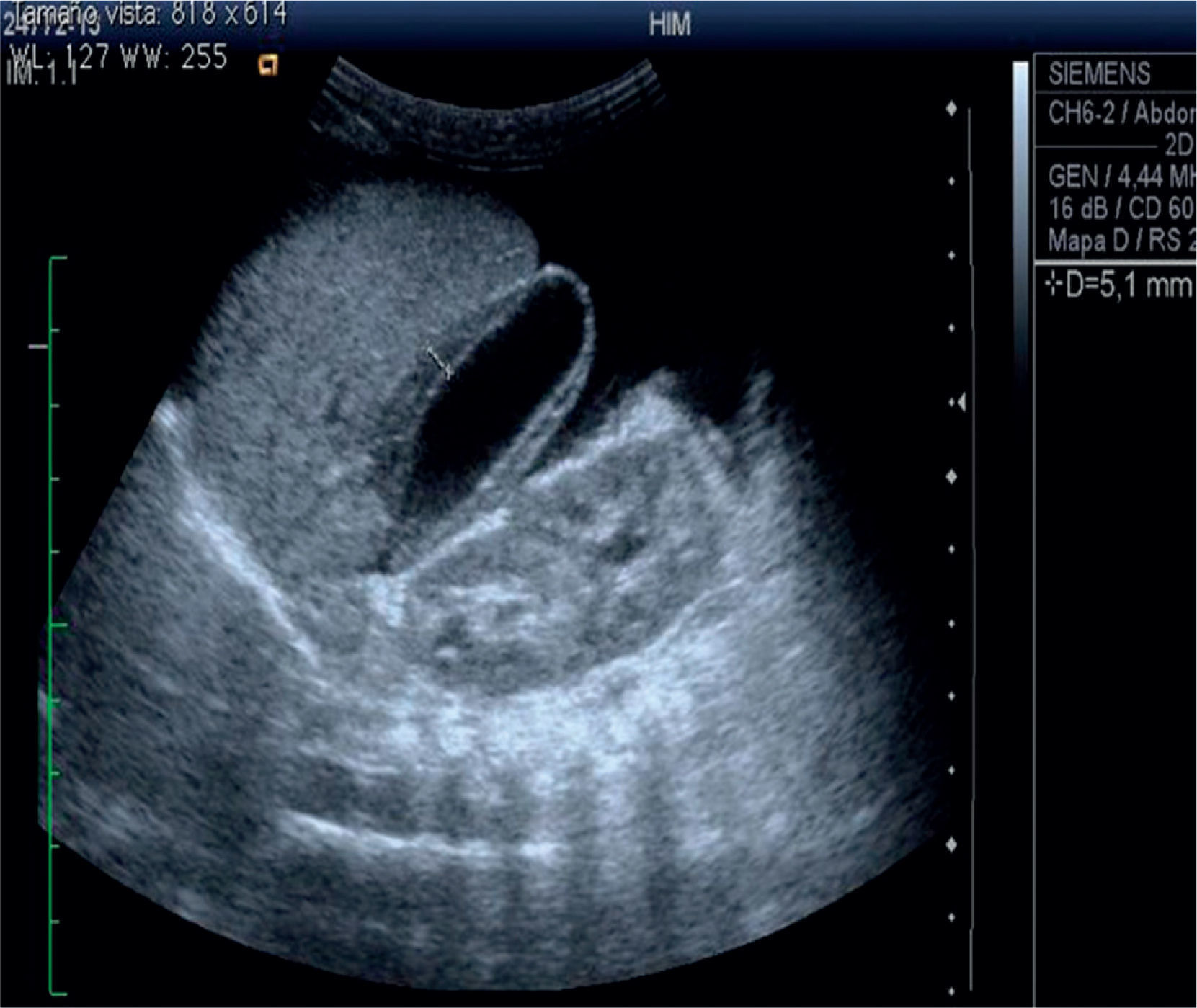
The abdominal ultrasound shows heterogeneous liver echogenicity, microlobulated edges, and a portal vein diameter of 4.8mm. Portal flow was hepatopetal of 26cm/s, there were collateral vessels with helical flow in the hilium. The hepatic artery had an IR of 0.9 and hepatic veins were permeable. The ultrasonogram conclusion was chronic liver damage, and portal hypertension with probable portal vein cavernomatous transformation (Figure 2).

Head computed tomography (CT) showed increased depth of sulci and fissures, increased compensatory volume of the ventricular system and an arachnoid cyst. There was no evidence of hemorrhage or ischemia.
3Case presentationThe syndromatic diagnoses were:
- •
Syndrome of portal hypertension, intrahepatic type: ascites, collateral vessels, ultrasonographic finding of suprahepatic and portal vein permeability.
- •
Cholestatic syndrome: jaundice and serum direct bilirubin greater than 2mg/dl.
- •
Systemic inflammatory response syndrome: fever, tachycardia, and leukocytosis.
- •
Encephalopathic syndrome: disturbance in the sleep pattern, irritability and decreased alertness, with no focal neurologic signs.
- •
Hemorrhagic syndrome: Based on bleeding from puncture sites, endotracheal tube, and gastric drainage, bleeding gums, low hemoglobin and hematocrit, thrombocytopenia and increased clotting times.
- •
Respiratory distress syndrome: Based on intercostal retractions, nasal flaring and thoracoabdominal dissociation.
- •
Abdominal compartment syndrome: Based on increased waist circumference and intra-abdominal pressure with impaired renal function.
Nosological diagnoses were:
- •
Hepatic encephalopathy based on neurological impairment and hyperammonemia.
- •
Hepatic impairment based INR> 1.5 without improvement after vitamin K administration, hypoglycemia, liver encephalopathy and hypoalbuminemia.
- •
Vitamin D insufficiency given vitamin D concentration of 21–29ng/dl.
- •
Sepsis based on inflammatory response syndrome, abdominal manifestations and positive blood cultures.
- •
Proximal tubulopathy given the glycosuria, hypophosphatemia, hypokalemia and hyperphosphaturia.
- •
Prerenal renal failure based on elevated serum creatinine, urea and FeNa of 0.6%
- •
Disseminated intravascular coagulation based on the hemorrhagic syndrome, thrombocytopenia, prolonged clotting times and decreased fibrinogen.
- •
Septic shock based on evidence of systemic inflammatory response, hypotension, cold extremities, weak pulses and need of norepinephrine infusion.
- •
Multiple organ failure: neurological, cardiovascular, respiratory, hematological, hepatic and renal dysfunction.
Regarding the etiology of liver failure, bile canalicular and structural anomalies were unlikely because absent acholic or hypocholic stools, or choluria. Common infectious causes of liver disease were ruled out, as serology was negative for hepatitis A, B and C, cytomegalovirus, Epstein-Barr virus, toxoplasma, rubella and HIV. Some less common etiologies that were not discarded were ECHO virus, Coxsackie virus, adenovirus, parvovirus and chicken-pox. Hypothyroidism was also ruled out.
Liver disease of rapid progression to liver failure, developmental delay, tubulopathy and bilateral nuclear cataract pointed to a metabolic disease. Fatty acid oxidation disorders were ruled out by ketonuria and normal BUN. A disorder of carbohydrate metabolism was unlikely since, while hemodynamically stable, there was no hypoglycemia, metabolic acidosis or hyperlactatemia. Galactose quantification, urine reducing substances and liver biopsy were not available.
Respiratory alkalosis, ketonuria and increased ammonia up to 100mmol/l suggested a defect in the urea cycle, but we could not confirm the deficiency of a specific enzyme. Furthermore, although amino acid profile was described as normal, succinyl acetone and urine organic acids were unavailable to rule out an aminoacidopathy.
As discussed above, the diagnostic possibilities were galactosemia, glycogenosis, tyrosinemia and an urea cycle disorder. Moreover, given the normal levels of gamma-glutamyltranspeptidase, other alternatives were impaired synthesis of bile acids or progressive familial intrahepatic cholestasis.
It is worth noting that this patient had also failure to thrive (height for age at birth of 98% reduced to 89% at 5 months of age), and also suffered severe malnutrition according to WHO criteria for patients aged 6 to 60 months (weight for height less than 70%).
This patient probably had spontaneous bacterial peritonitis on admission to the emergency room since she had a waist circumference 7cm larger than in the first assessment, progressive jaundice, increased direct bilirubin and prolonged clotting times. To corroborate this diagnosis a paracentesis and peritoneal fluid culture were needed.
Hyponatremia in patients with liver disease and ascites is usually dilutional, because of renal retention of sodium and water. The main indications for hyponatremia correction are neurological symptoms and serum sodium less than 120mEq/l. Fluid restriction is the most important action to increase serum sodium concentration. When hypokalemia is associated to hyponatremia, as in this case, it should be corrected.
Refractory ascites is defined as the one which cannot be mobilized or reappears early after its treatment. It occurs in 5–10% of patients, and is subdivided into two groups: diuretic resistant ascites and diuretic intractable ascites. This patient was in the latter group, since she progressed to kidney failure that contraindicated treatment with diuretics at high doses.
During her evolution, the patient had increased creatinine with no improvement after volume expansion with crystalloid fluids and albumin infusion. Hepatorenal syndrome should be suspected in cirrhotic patients with ascites who do not have renal parenchymal disease and have urinary sodium of 6mmol/l. However, the patient did not meet the criteria for diagnosis because she had received nephrotoxic drugs and serum creatinine was below 1.5mg/dl.
Final diagnoses were:
- •
Liver disease, probably secondary to an inborn error of metabolism
- •
Intrahepatic portal hypertension
- •
Acute liver failure
- •
Grade III hepatic encephalopathy
- •
Intractable ascites
- •
Sepsis with an abdominal origin
- •
Severe acute malnutrition
- •
Growth retardation of moderate intensity
- •
Developmental delay
This was a necropsy of an infant with generalized jaundice, portal hypertension, abdominal distension due to ascites, collateral vessels in thorax and abdomen, and reduced muscle mass. Peritoneal clear fluid was found (950ml) in the abdominal cavity. There was an epicardial bleeding.
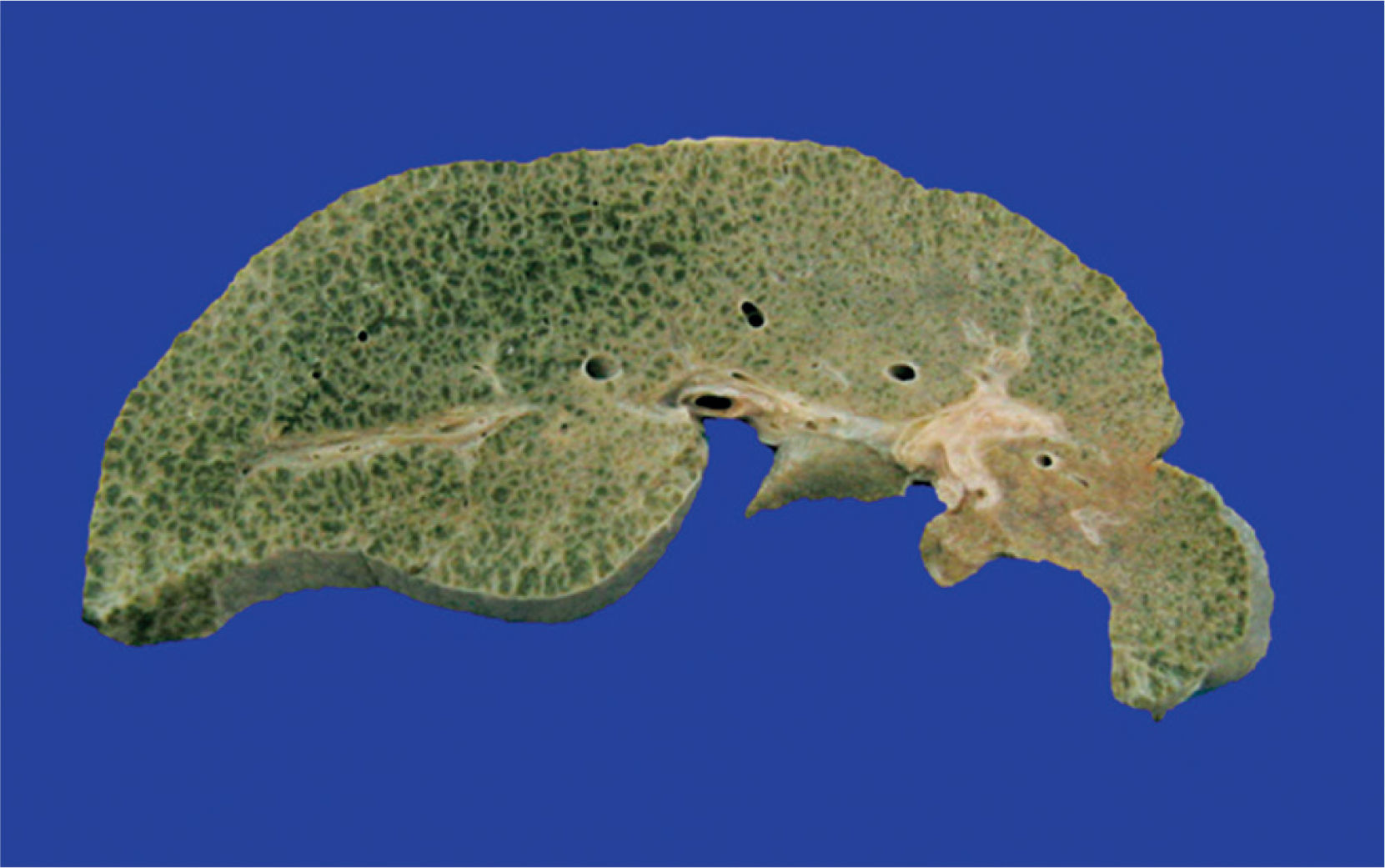
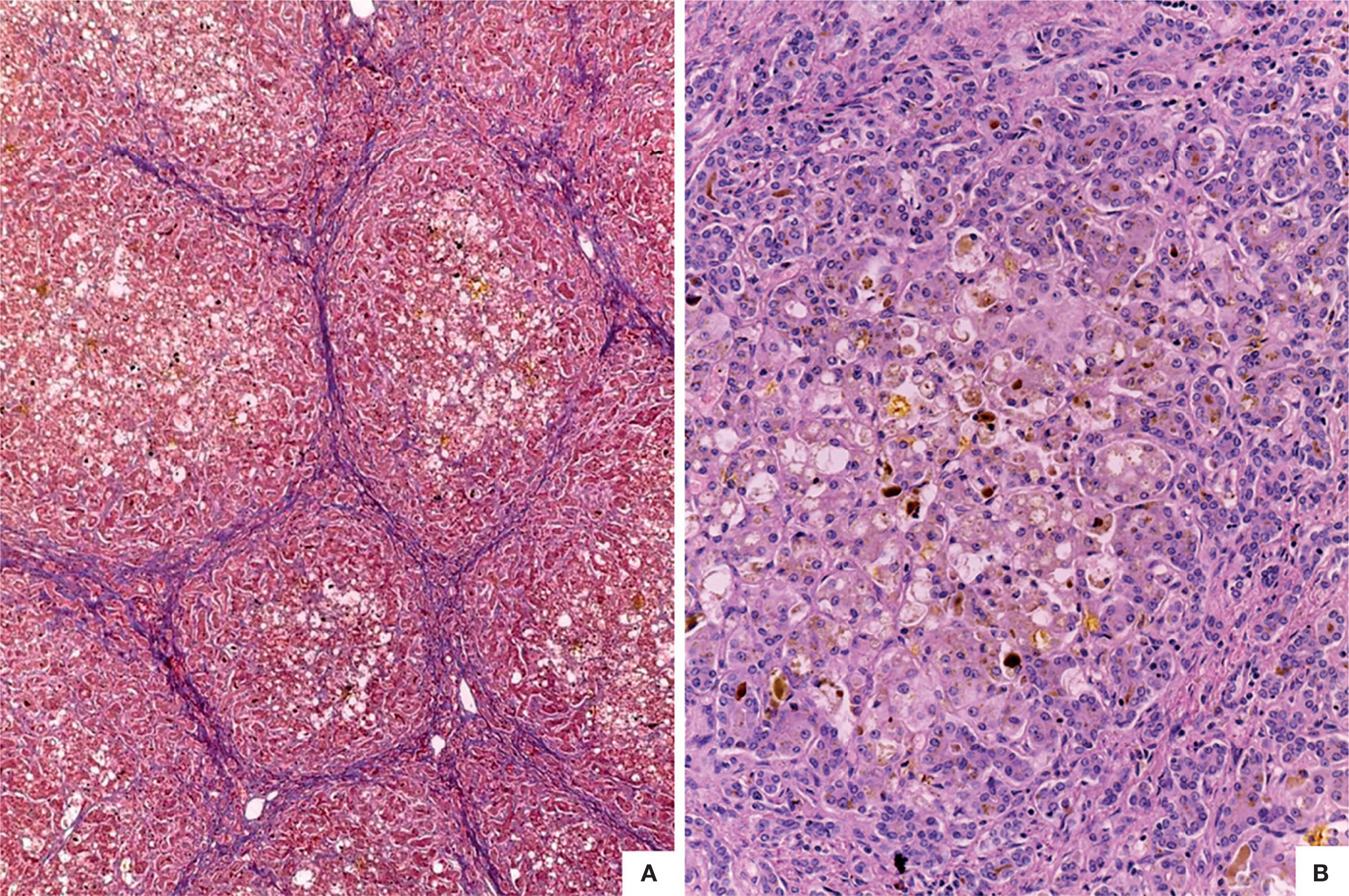
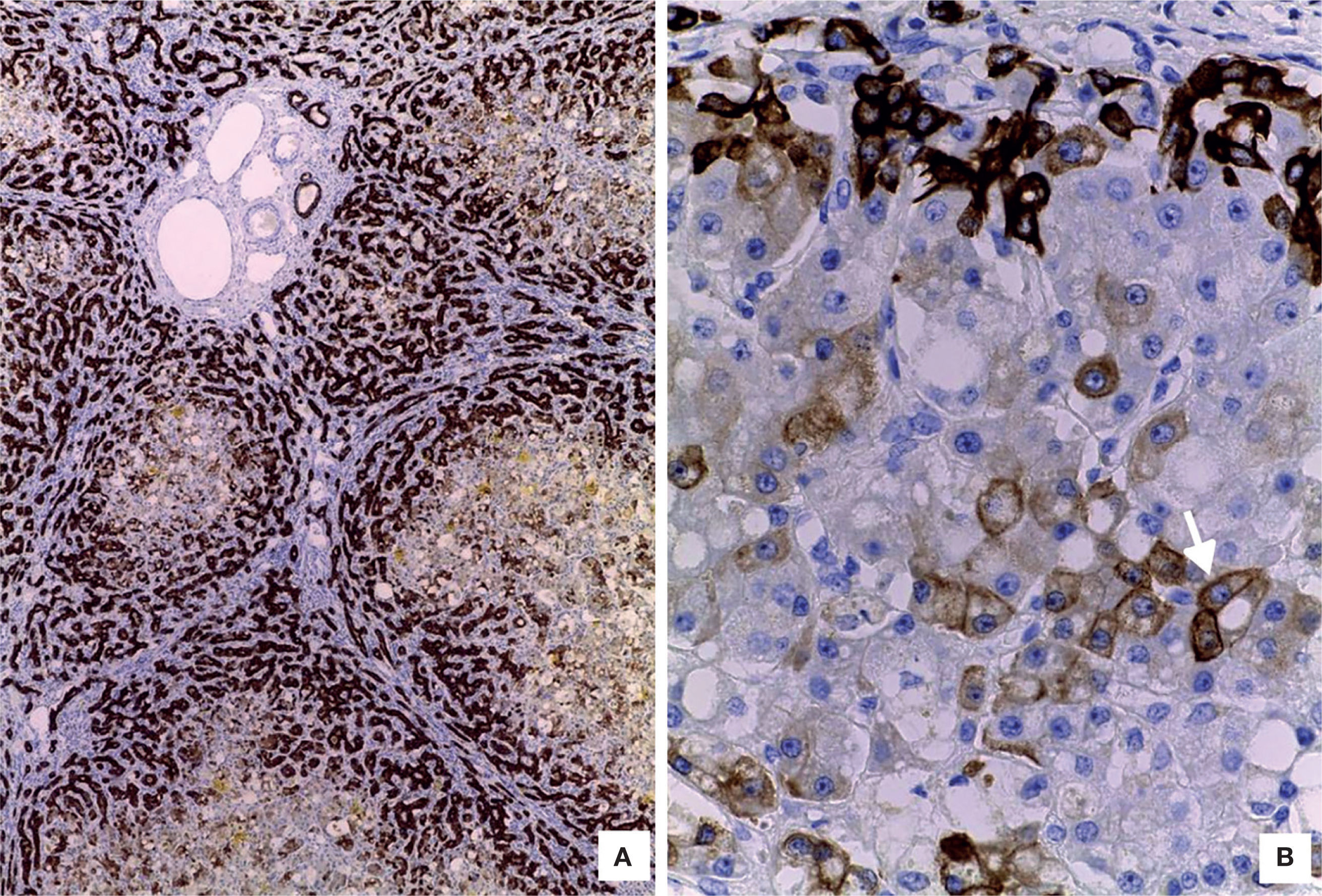
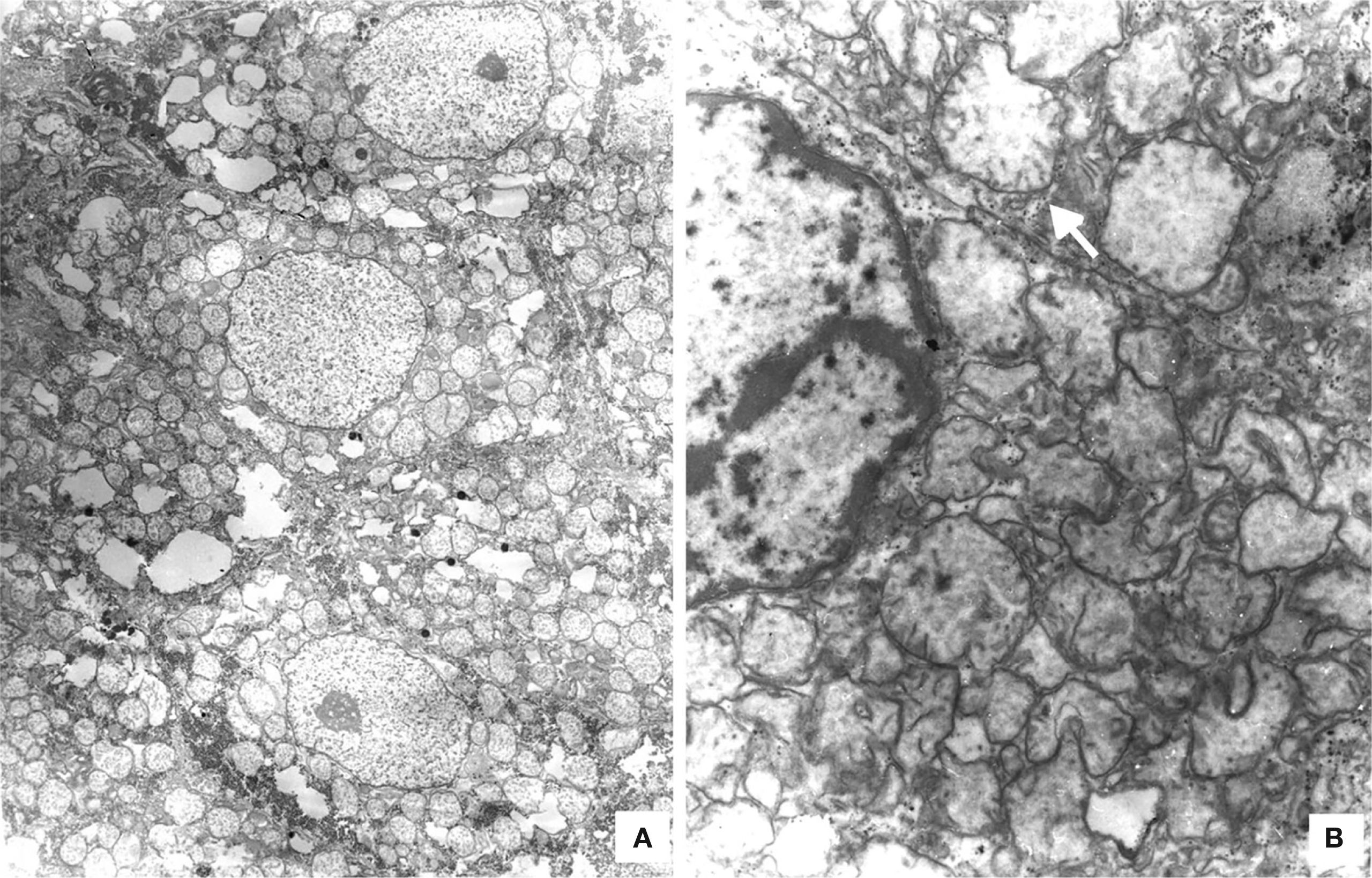
The liver was enlarged, green colored an with a micronodular surface; transversal serial sections showed regenerative nodules with a diameter smaller than 3mm, multifocal areas of recent necrosis and dilatation of intrahepatic portal veins.(Figure 3). Histopathological examination showed micronodular liver cirrhosis. Regeneration nodules were surrounded by thin layers of fibrosis and numerous cholangioles, some with bile plugs in their lumen. There was evidence of recent and past extensive necrosis of hepatocytes, mainly centrilobular, associated with macrovesicular steatosis and canalicular hepatocellular cholestasis (Figure 4). There were not hypereosinophilic hepatocytes due to the intense and chronic damage. The findings were consistent with those described in depletion of mitochondrial DNA (mtDNA) syndrome1. Immunohistochemmical staining for cytokeratin-7 (CK7) showed intense canaliculi proliferation in the periphery of regeneration nodules and an abnormal expression of CK7 in hepatocyte cytoplasm highlighting the extensive hepatocytes loss (Figure 5). Iron storage was noticed in hepatocytes and Kupffer cells. Transmission electron microscopy showed hepatocytes with enlarged cytoplasm and an increased number of mitochondria which displaced the nucleus and other organelles. These atypical mitochondria had irregular shapes and sizes, with an increased matrix volume of granular appearance and cristae decreased in number and displacement to the periphery (Figure 6). In other areas there were bile deposits and myelin-like structures which pointed to the diagnosis of a mitochondrial disease. Some authors suggest that these ultrastructural findings are characteristic of mtDNA depletion syndrome. Patients with the mutation MPV17 located on 2p24.1 show clinical and histopathological findings similar to this patient.1-4 However, it is necessary to perform molecular biology tests to confirm this mutation.

. B. Hepatocytes with cytoplasmic vacuolation and cholestasis. There is cholangiole proliferation in the periphery of regeneration nodules (HE 100X).")
. Cholangioles highlighted by cytoplasmic expression of CK7 (IHC).")
.")
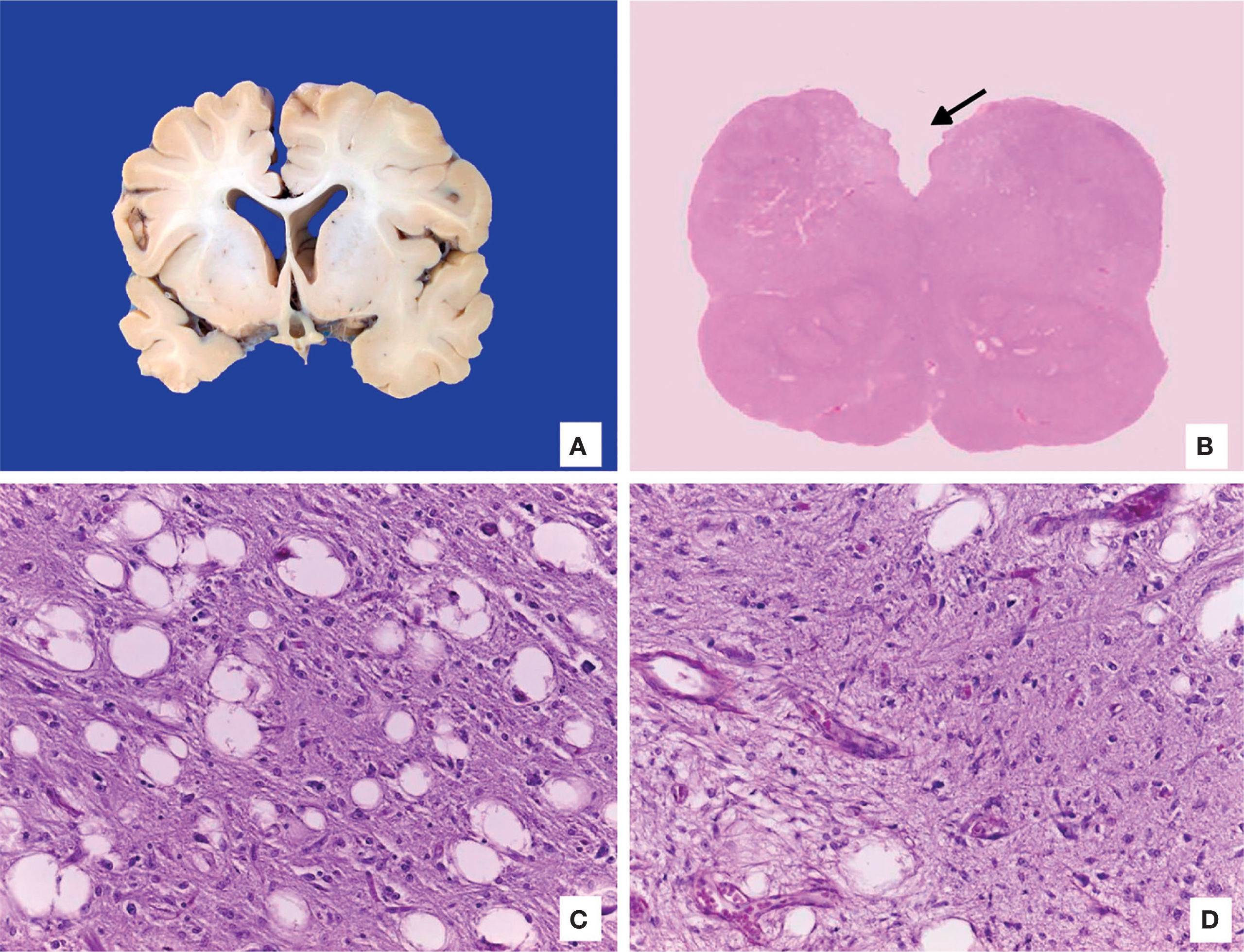
The brain was atrophic with reduced weight (750g vs. 810g), deep sulci and narrow gyri, exvacuo dilatation of lateral ventricles and decreased white matter. No alterations in gray basal ganglia or cerebellum were observed. Microscopic examination showed the presence of Alzheimer type 2 astrocytes in the cortex, which were related to the patient’s liver failure. Medulla had areas of encephalomalacia with vascular proliferation and myelin vacuolation (Figure 7).
. C. Neuropil vacuolation and loss of myelin. D. Vascular proliferation.")
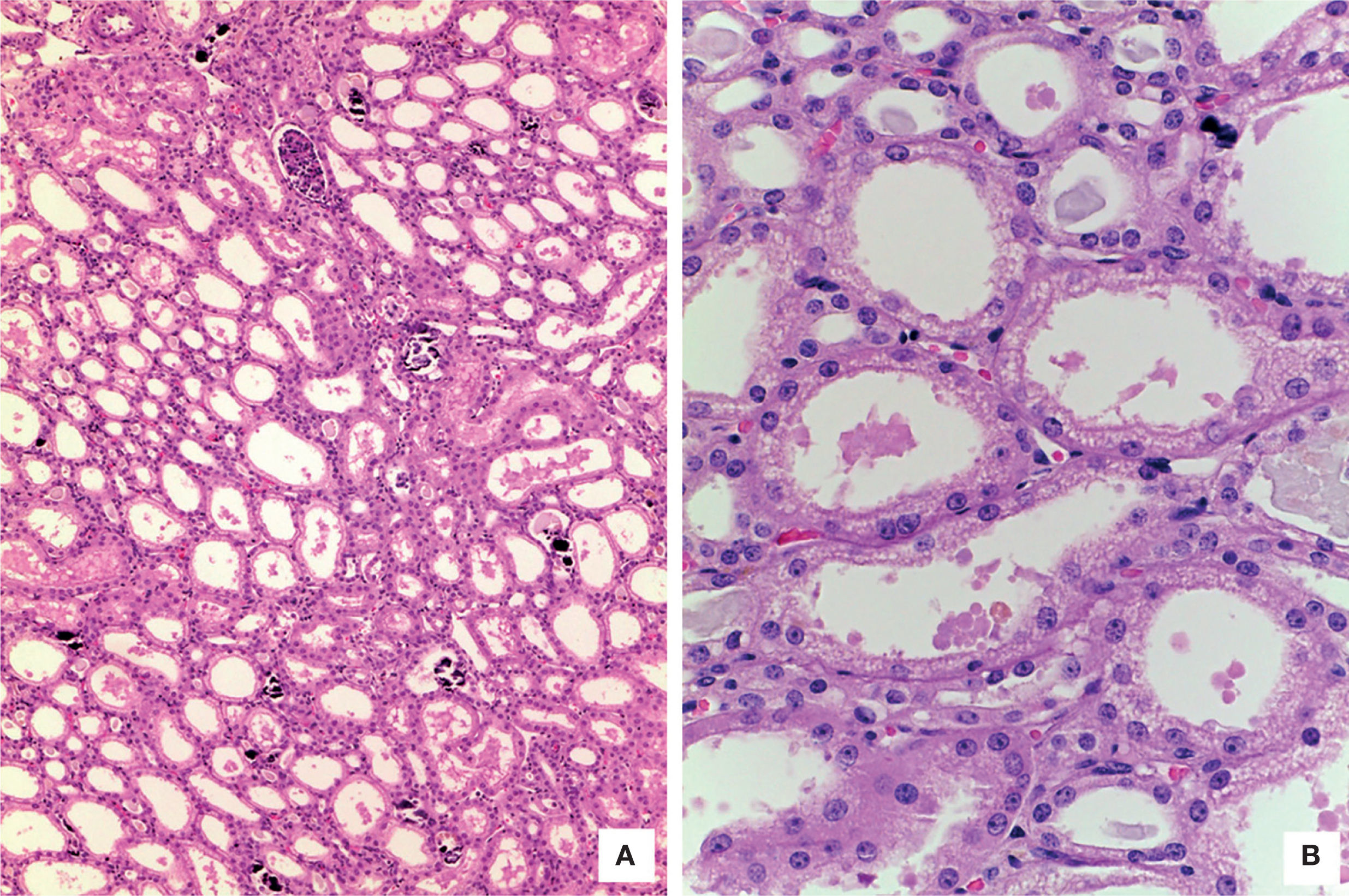
The kidneys showed medullary congestion; as an independent finding, the right kidney had double collecting system. Histological sections showed lobulated parenchyma, extensive nephrocalcinosis and normal glomeruli. The most important finding was proximal tubules dilatation. Epithelial cells showed large vacuolated cytoplasm with preserved luminal border. Ultrastructural findings were similar to those found in hepatocytes’ mitochondria and were responsible of proximal tubulopathy (Figure 8).5 Skeletal muscle was morphologically normal.
. B. Proximal tubules with cytoplasm vacuolation and lumen dilatation (HE 400X).")
The most frequent type of mtDNA depletion syndrome is the hepatocerebral, which starts in the neonatal period, is progressive and lethal by the first year of life because of liver failure (Table 4). This syndrome is considered as a defect in mtDNA maintenance due to an impaired communication between the nuclear and mitochondrial DNA.2
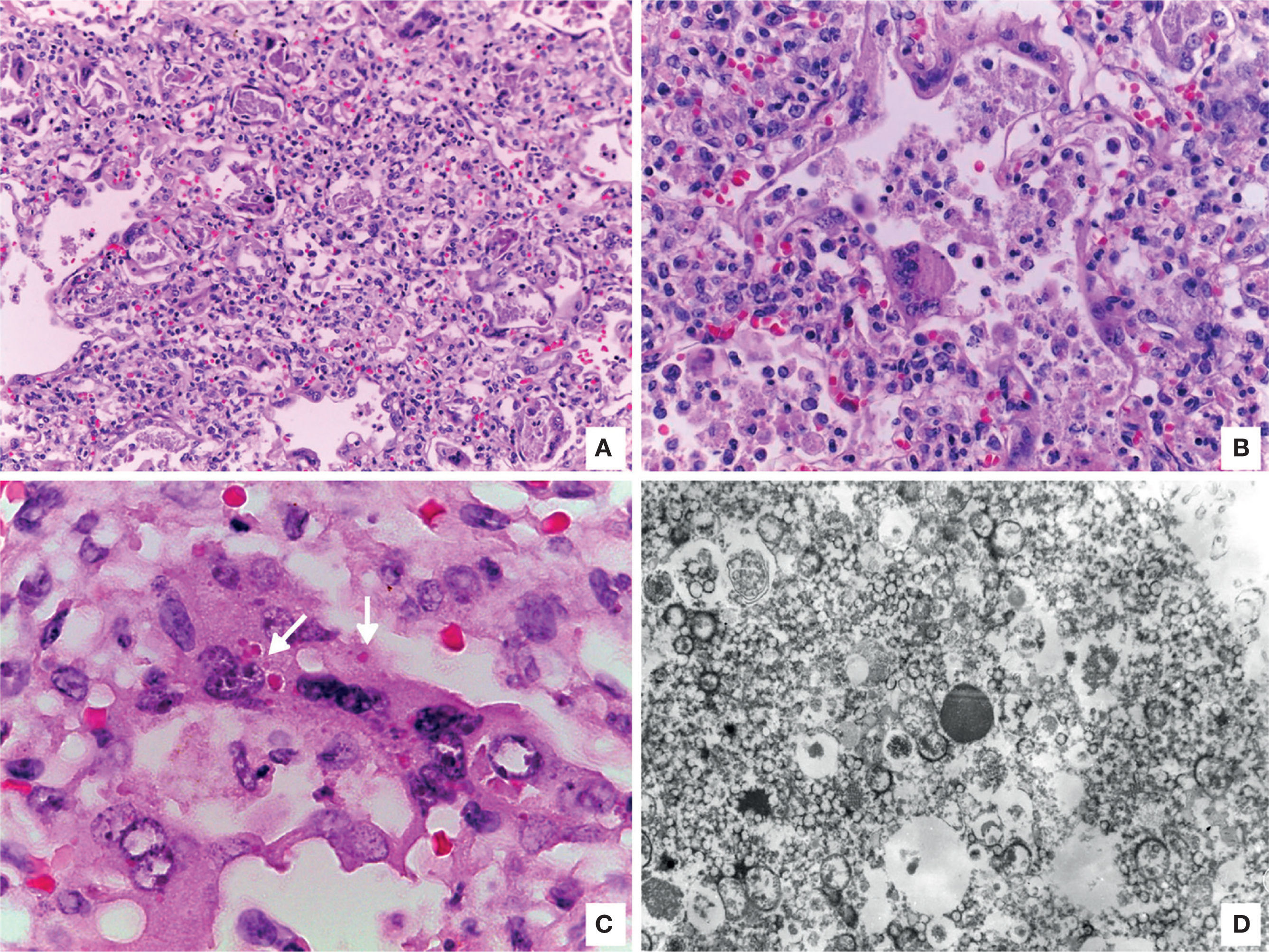
Other findings were viral pneumonia with diffuse alveolar damage, numerous giant cells, some with intracytoplasmic eosinophilic inclusions surrounded by a clear halo; electron microscopy analysis of inclusions showed electrodense filamentous structures consistent with respiratory syncytial virus (Figure 9). The myocardium showed colliquative myocitolysis, cardiomyocyte hypertrophy, and morphological data of multiorganic shock.
. D. Electronic microscopy showing filamentous inclusions compatible with respiratory syncytial virus.")
A. Lung with viral pneumonia signs. B. Alveoli are lined by giant multinucleated cells and there is lymphocytic infiltrate in alveolar septa. C. Giant multinucleate cell with eosinophilic cytoplasmic inclusions (arrows). D. Electronic microscopy showing filamentous inclusions compatible with respiratory syncytial virus.
Mitochondrial DNA depletion syndrome, type hepatocerebral with proximal tubulopathy.
4.2Concomitant alterations- •
Hepatomegaly 800g vs. 276g
- •
Ascites 950ml
- •
Micronodular cirrhosis
- •
Hepatocellular canallicular and ductal cholestasis
- •
Macro and microvesicular hepatic steatosis and mild hemosiderosis
- •
Ultrastructural mitochondrialchanges in hepatocytes
- •
Cerebral atrophy 750g vs. 810g
- •
Moderate medullaencephalomalacia
- •
Bilateral nephromegaly
- •
Proximaltubulopathy with epithelial vacuolation and mitochondrial ultrastructural changes.
- •
Extensive nephrocalcinosis
- •
Giant cell viral pneumonia by respiratory syncytial virus.
- •
Acute diffuse alveolar damage
- •
Grade B pulmonary vascular disease
- •
Cardiomegaly 50g vs. 41g
- •
Myocardic colliquative myocytolysis
- •
Abnormal lung lobation
- •
Double right collecting system
This condition could have been diagnosed with a liver biopsy, since there were characteristic histopathological and ultrastructural findings.
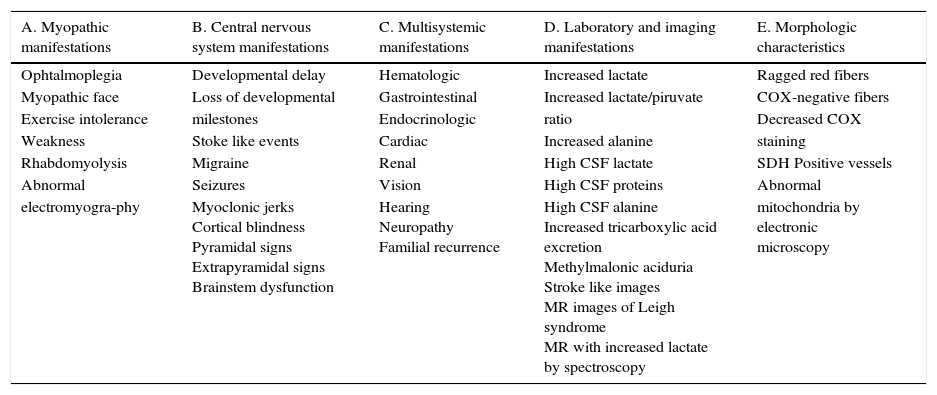
5Final comments5.1Department of GeneticsThe human genome can be divided into two groups: the nuclear genome and the mitochondrial genome. The nuclear genome has approximately 26 thousand genes, while the mitochondrial has 37 genes. Thirteen mitochondrial genes encode proteins that are part of the respiratory chain, the site of ATP production by oxidative phosphorylation.1 There is an important communication and interaction between the nuclear and mitochondrial genomes. There are some genes in nuclear DNA which encode proteins responsible of maintenance, replication and transcription of mtDNA; thus, the proper function of both genomes is necessary to mtDNA maintenance, and therefore, for ATP generation.6 Mitochondrial diseases are a group of multisystemic disorders, which frequently affect the central and peripheral nervous system; they are caused by different mutations located both in the mitochondrial and nuclear genomes. The prevalence of these diseases is 1 in 5,000 individuals.7 Clinical manifestations may occur in any organ or tissue, at any age and with any inheritance mechanism (Table 4). Due to the complexity of these diseases, diagnostic criteria and diagnostic probability scales have been developed in order to establish degrees of certainty for the existence of a mitochondrial dysfunction, as the modified Wolf scale (Table 5).8
Modified Wolf scale.
| A. Myopathic manifestations | B. Central nervous system manifestations | C. Multisystemic manifestations | D. Laboratory and imaging manifestations | E. Morphologic characteristics |
|---|---|---|---|---|
| Ophtalmoplegia | Developmental delay | Hematologic | Increased lactate | Ragged red fibers |
| Myopathic face | Loss of developmental | Gastrointestinal | Increased lactate/piruvate | COX-negative fibers |
| Exercise intolerance | milestones | Endocrinologic | ratio | Decreased COX |
| Weakness | Stoke like events | Cardiac | Increased alanine | staining |
| Rhabdomyolysis | Migraine | Renal | High CSF lactate | SDH Positive vessels |
| Abnormal | Seizures | Vision | High CSF proteins | Abnormal |
| electromyogra-phy | Myoclonic jerks Cortical blindness Pyramidal signs Extrapyramidal signs Brainstem dysfunction | Hearing Neuropathy Familial recurrence | High CSF alanine Increased tricarboxylic acid excretion Methylmalonic aciduria Stroke like images MR images of Leigh syndrome MR with increased lactate by spectroscopy | mitochondria by electronic microscopy |
CSF, cerebrospinal fluid; MR, magnetic resonance; COX, cytochrome C oxidase; SDH, succinate dehydrogenase.
Recently, new molecular techniques for mutation identification have been implemented, such as second generation sequencing, since mtDNA and nuclear DNA mutations cause similar signs and symptoms. The mtDNA depletion syndrome is a heterogeneous group of disorders caused by the decrease in the number of copies of mtDNA because of mutations in nuclear genes encoding proteins responsible for replication and maintenance of mtDNA. They have an autosomal recessive inheritance pattern and genetic heterogeneity; i.e., mutations in different genes can cause the mtDNA depletion syndrome.9,10
In summary, mtDNA depletion syndrome should be considered when a newborn or infant presents with hypoglycemia, hyperlactatemia and hyperbilirubinemia. Ultrastructural mitochondrial findings in a liver biopsy may orient the diagnosis of mtDNA depletion syndrome. However, molecular biology studies are required for an accurate diagnosis.
Ethical disclosureRight to privacy and informed consentThe authors declare that no patient data appear in this article.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Protection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Conflict of interestThe authors declare no conflict of interest of any nature.
Please cite this article as: Villalpando Carrion S, et al. Lactante con ictericia progresiva, cirrosis y tubulopatía proximal. Bol Med Hosp Infant Mex. 2016;73:129-38.