


Sarcoidosis is a systemic disease of unknown etiology that rarely occurs in children. It usually affects the lungs; however, it may involve various organs. It occasionally affects the general condition, and causes fever, hepatomegaly and splenomegaly.
Case reportWe report the case of a twelve-year-old adolescent with late-onset childhood sarcoidosis which diagnosis was confirmed by lymph node histopathological study. The patient presented general condition, hypercalcemia, erythema nodosum, severe lung disorders, lymphadenopathy, hepatomegaly and testicular mass. He received treatment with steroids, with excellent clinical response.
ConclusionsWe highlight the importance of considering the diagnosis of sarcoidosis in patients with hepatomegaly, lymphadenopathy, diffuse lung damage, erythema nodosum, testicular mass and hypercalcemia, as well as the need for a multidisciplinary approach to assess multiple organ involvement and the early beginning of steroid treatment in order to prevent the progression of the disease.
La sarcoidosis es una enfermedad sistémica de etiología desconocida que raramente se presenta en la infancia. Generalmente afecta los pulmones; sin embargo, puede involucrar diversos órganos. Ocasionalmente afecta el estado general, y origina fiebre, hepatomegalia y esplenomegalia.
Caso clínicoSe presenta el caso de un adolescente de doce años de edad con sarcoidosis infantil de inicio tardío, cuyo diagnóstico fue confirmado con un estudio histopatológico de ganglio linfático. El paciente cursó con afección general, hipercalcemia, eritema nodoso, alteraciones pulmonares graves, adenopatías, hepatomegalia y masa testicular. Recibió tratamiento con esteroides, con excelente respuesta clínica.
ConclusionesSe resalta la importancia de considerar el diagnóstico de sarcoidosis en los pacientes con hepatomegalia, adenopatías, daño pulmonar difuso, eritema nodoso, masa testicular e hipercalcemia, así como la necesidad del abordaje multidisciplinario para valorar el compromiso orgánico múltiple y el inicio oportuno de la terapia con esteroides, con el fin de evitar la progresión de la enfermedad.
Sarcoidosis is a chronic systemic disease of unknown etiology and worldwide distribution which is usually diagnosed in adults. Sarcoidosis in childhood is very rare.
Lungs are the most frequently affected organs; however, the disease can involve other organs such as eyes, skin, lymph nodes and joints. Less often it involves the nervous system, heart and urogenital tract, causing nephrolithiasis and a testicular mass; in some cases, fever of unknown origin with splenomegaly and hepatomegaly are observed.
The diagnosis of sarcoidosis is made by exclusion of other diseases. Therefore, once there is clinical suspicion, a biopsy of the organs involved shows, as a characteristic histopathological finding, the presence of noncaseating epithelioid granulomas.1
The first descriptions of sarcoidosis were made in Europe in the late nineteenth century. In 1877, in England, J. Hutchinson studied a patient with chronic skin lesions, arthritis and chronic renal failure, and named the skin findings papillary psoriasis. In France, in 1889, E. Besnier also described the skin lesions and named them lupus pernio. In 1899, in Denmark, C. Boeck labeled the skin histological lesions with the term sarkoid because of its similarity with sarcoma. In 1914, in Sweden, N. J. Schaumann described the systemic presentation of the disease; likewise, he pointed out that both Besnier’s lupus pernio and Boeck’s sarkoid were manifestations of the same disease, as the tissues affected in these patients showed granulomas which he called benign lymphogranulomatosis to differentiate them from Hodgkin’s malignant granuloma. The neurologic involvement in sarcoidosis was reported by C. Heerfordt in 1923 who described patients with uveo-parotid fever and lesion of cranial nerves.
The acute pulmonary form of sarcoidosis accompanied of mediastinal lymphadenopathy and erythema nodosum, arthritis and uveitis was described in Sweden in 1953 by S. Löfgren.
For more than 130 years, most of the studies on sarcoidosis have been performed in adults. However, pediatric cases have been reported since 1923. The condition was known as Besnier-Boeck-Schaumann disease until 1958 when the Sarcoidosis World Congress was carried out in London and the term sarcoidosis was generalized.2,3
Nowadays, accordingly to the international consensus established by the American Thoracic Society, the European Respiratory Society and the World Association for Sarcoidosis and Other Granulomatosis, sarcoidosis is considered to be a systemic granulomatous disease of unknown etiology, which usually affects adults, and is very rare in children.4,5
The incidence and severity of sarcoidosis vary in different regions of the world and in different ethnic groups probably due to variations in environmental exposures, the prevalence of HLA alleles and other genetic factors. Scandinavia, England, the United States and Japan have the highest prevalence of the disease; in Sweden, the morbidity rate in the general population is 64/100,000 and in the United States, 35/100,000.
There are very few epidemiological data on children. The rate of morbidity of childhood sarcoidosis is 0.29/100,000; however, this rate varies from 0.06/100,000 in children under 4 years to up to 1.02/100,000 in adolescents aged 14 to 15 years.6-8
Regarding mortality from sarcoidosis, during the period 1999-2010 the National Center for Health Statistics reported sarcoidosis as a cause of death in 10,348 of 29,176,040 deaths which represents a rate of 2.8/1,000,000 inhabitants. However, mortality in the African-American population was 12 times higher than in Caucasian population.9
In Mexico, the incidence of sarcoidosis is low, probably due to genetic factors or underreporting of cases. Therefore, there are no studies on the epidemiology of childhood sarcoidosis since only thirteen cases have been published in the last 20 years, including the case of a teenager with lung disease published by the National Institute of Respiratory Diseases and another report from the National Institute of Pediatrics about two cases of early-onset cutaneous sarcoidosis.10-12
Sarcoidosis is a chronic inflammatory disease that results from the action of an environmental agent that triggers an initial immune response of T-helper cells type 1(Th1) and leads to the development of noncaseating granulomas with systemic involvement in genetically susceptible individuals.
According to epidemiological findings of the multicenter ACCESS (A Case Control Etiologic Study of Sarcoidosis), some environmental conditions are associated with an increased risk for developing sarcoidosis, generating antigenic stimuli, which act as a trigger of the process. Some of the environmental conditions studied are organic materials (ragweed, pine leaves and seeds), inorganic materials (silica, beryllium, zirconium, titanium, aluminum and fiberglass) and microorganisms (Mycobacterium tuberculosis, Propionibacterium acnes, Brucella, Borrelia, Leptospira, Mycoplasma, Leishmania and Schistosoma).
Although an infectious agent has not been conclusively identified (by culture or by ribosomal RNA markers in tissue from biopsies), the hypothesis of a microbiological agent is the most accepted because there is clinical and epidemiological evidence of transmissibility of sarcoidosis. This observation comes from transplant patients who have developed the disease after tissue or organ transplantation from donors with sarcoidosis. Besides, it has been reported the development of sarcoidosis in the transplanted lung of a patient suffering from the disease; moreover, animals implanted with tissue from affected patients develop granulomas.13-18
The existence of a predisposing genetic factor explains the higher incidence of the disease in relatives of patients with sarcoidosis, as well as differences in the prevalence and clinical course of the disease in different ethnic groups.
Recent research with molecular biology techniques has shown that genetic alterations associated with sarcoidosis are located in the major histocompatibility complex (MHC), on the short arm of chromosome 6, in histocompatibility antigens HLA I, such as HLA-B7 and HLA-B8, as well as in alleles of HLA class II such as HLA-DR5, HLA-DR6, HLA-DR8 and HLA-DR9, which are related to high risk in Asian population. Likewise, genetic alterations are found in HLA-DRB1 in the African American population. In European population, HLA-DR14 and HLA-DR15 are related to chronic sarcoidosis, HLA-DR3 with the acute form, and HLA-DR17 with self-limited presentations of sarcoidosis.
The immune mechanisms that cause sarcoidosis are not completely known but it is assumed that macrophages do not adequately recognize and present antigens to T lymphocytes. However, patients with sarcoidosis do not have other evident manifestation of a cellular or humoral immunodeficiency.
The process begins when the antigen phagocyted by the macrophage and the dendritic cell are presented on the MHC site. This enables the T cell to locate and bind to the MHC-II complex, and consequently to be activated and have clonal expansion, demonstrated by the increase in T cell receptor (TCR) mRNA, and by the presence of markers of antigen-specific T cell activation, such as CD69, glycoprotein 240 and the very late antigen-1 (VLA-1).
Afterwards, T-CD4 lymphocytes differentiate to a Th1phenotype, and release cytokines such as interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), transforming growth factor-β (TGF-β), IL-1β, IL-2, IL-12, IL-15 and IL-18, as well as several chemokines such as macrophage inflammatory protein 3α (MIP3α), interferon-inducible protein-10 (IP-10), monocyte chemotactic protein-1 (MCP-1) and CCL5 or RANTES (regulated on activation, normal T cell expressed and secreted). These chemokines maintain cell recruitment at sites of granuloma formation by their chemotactic effect.
T-regulatory cells from patients with chronic active sarcoidosis have altered function which leads to the persistence of inflammatory process and progression of granulomas. By contrast, patients with Löfgren syndrome, which is a form of acute sarcoidosis with a high rate of remission, have increased T-regulatory cells which reduce lymphocyte proliferation and cytokine production.
In some patients, the initial Th1-type response is replaced by a predominantly T-helper cell type 2 (Th2) response, characterized by a decrease in IFN-γ, persistence of TNF-¿and TGF-β activities, production of angiotensin converting enzyme (ACE), neopterin and IL-8. These proteins reduce the formation of granulomas but, on the other hand, lead to the development of fibrosis due to the activity of some macrophage cytokines, like type1-insulin growth factor (IGF-1), platelet growth factor, IL-4 and IL-13 which generate fibroblast activation and collagen and fibronectin deposition in the extracellular space. The cause of progression to this fibro-proliferative form is unclear, but it may involve the loss of apoptotic mechanisms, loss of regulatory response, or persistence of an antigen that cannot be recognized or properly processed, which causes fibrous scarring and repair of the affected tissue with chronic and irreversible damage.19-24
Vitamin D deficiency has been shown to lead to an increased risk of sarcoidosis, as the antigen presenting cells (macrophages, monocytes and dendritic cells) have vitamin D receptors. This vitamin inhibits macrophage activation induced by IFN-γ and decreases the macrophage MHC II antigen presenting activity; so, it is supposed that this deficiency leads to altered regulation of Th1 cells, allowing a persistent immune response.25-27
Furthermore, it has been found that activated macrophages are capable of producing active vitamin D3 (calcitriol or 1,25-dihydroxycholecalciferol) and parathyroid hormone related peptide (PTHrP), which could contribute to hypercalcemia and hypercalciuria frequently present in this condition.28,29
One of the most remarkable findings in sarcoidosis is an immunological paradox. This paradox is characterized by intense cellular immune response in the affected organs in contrast to a situation of peripheral immunological anergy, observed as a lack of response to PPD and other intradermal tests. The likely explanation for this phenomenon is the presence of lymphopenia in peripheral blood, and especially to the activity of suppressor T CD8+ lymphocytes.
Another phenomenon which is also subject of interest and controversy is the formation of granulomas in the skin of patients with sarcoidosis four to six weeks after the application of sarcoid tissue extract in the Kveim-Siltzbachtest (used in the past for diagnostic purpose).30,31
The histological finding characteristic of sarcoidosis is the presence of noncaseating epithelioid granulomas diffusely scattered in different tissues that mainly affect lymph nodes.
The granulomas of sarcoidosis have compact appearance with well-defined borders. They may be at different stages of development, ranging from highly cellular granulomas to structures of decreased cellularity with fibrosis or progressive hyalinization; besides, they do not have central necrosis neither foreign bodies, in contrast to granulomas caused by mycobacterial and fungal infections, or infestations by systemic metazoans.
The typical sarcoidosis granuloma has two characteristic zones described below:
- 1.
The central zone or follicle is a dense cluster of epithelioid cells, accompanied by lymphocytes, macrophages, Langhans multinucleated giant cells, mast cells and fibroblasts. In 60% of the cases, star shaped structures (asteroid bodies) and lamellar PAS+ structures of 1–15 microns in diameter called Schaumann bodies can be observed. Immunohistochemical staining shows that the central zone of an active granuloma has macrophages in various stages of activation and differentiation. This zone is surrounded by CD4+ T cells intercalated with a small number of CD8+ T cells and B cells.
- 2.
The peripheral zone is formed by a ring of lymphocytes, monocytes and fibroblasts. T-regulatory CD3+/CD4+/CD25+/Foxp3+ cells accumulate in this outer zone in addition to CD8+ T cells and fibroblasts, which leads to fibrosis when granulomas activity decreases.
In the chronic form of the disease, granulomas may be encapsulated by a fibrous halo or may be replaced by scars of fibrous and hyaline tissue.32-34
The clinical presentation of sarcoidosis in children varies as it depends on the extent of the disease and the organs involved. In most cases of childhood sarcoidosis, multiple organs are affected with a diffuse inflammatory reaction that causes systemic symptoms such as fever, fatigue, hyporexia, nausea and weight loss, in addition to the specific signs and symptoms arising from dysfunction of each affected organ.
Sarcoidosis in childhood can occur in two clinical forms:
- 1.
Early onset sarcoidosis or Blau syndrome: It occurs before age five sporadically or in a familiar cluster. It is associated with NOD2/CARD15 gene located on chromosome 16. In 75% percent, the cases present a clinical triad characterized by polyarthritis, uveitis and rash, whereas the remaining 25% are accompanied by other organs signs.
- 2.
Late onset sarcoidosis: It develops in children older than 5 years and resembles the adult clinical form. It is not associated with mutations in the NOD2/CARD15 gene and it is characterized by fever, multiorgan involvement, especially lung, skin, nervous system, eyes, kidneys, joints, lymph nodes, liver and spleen.35-36
The lung is involved in over 90% of cases; however, the clinical spectrum of the disease is wide. Most cases occur in an acute form, with malaise and non-specific respiratory symptoms such as dry cough, dyspnea and airway hyperresponsiveness, but the disease can start insidiously and with minimal symptoms. The condition is mainly located in the pulmonary interstitium, with enlarged hilar, tracheobronchial and mediastinal lymph nodes.
Approximately 70% of cases resolve spontaneously, but the remaining 30% evolves to chronicity and cause irreversible changes in lung parenchyma with fibrosis and formation of pneumatoceles that lead to chronic respiratory failure and death.
Spirometry and plethysmography show a restrictive pattern, coincident with pulmonary dysfunction, and are useful to assess disease progression and response to treatment.
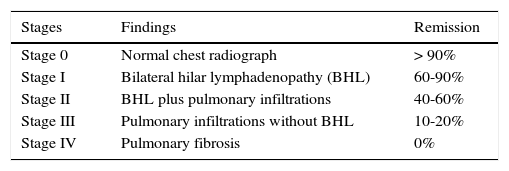
Sarcoidosis may be stratified in four radiographic stages, which guide the clinician to choose the treatment plan and to make a prognosis. In addition, radiographic imaging allows the follow up of the disease; the probability of remission decreases according to the stage of evolution of the lesions. This is, while radiographic resolution in the first three stages of the disease is feasible, during stage IV, which is considered the end stage, radiographic lesions are already irreversible37 (Table 1).
Chest radiographic staging*
| Stages | Findings | Remission |
|---|---|---|
| Stage 0 | Normal chest radiograph | > 90% |
| Stage I | Bilateral hilar lymphadenopathy (BHL) | 60-90% |
| Stage II | BHL plus pulmonary infiltrations | 40-60% |
| Stage III | Pulmonary infiltrations without BHL | 10-20% |
| Stage IV | Pulmonary fibrosis | 0% |
The incidence of ocular involvement varies from 30 to 70%. The characteristic lesion is uveitis. However, conjunctiva, sclera, crystalline and lacrimal glands may also be affected, resulting in cataract, glaucoma and dry keratoconjunctivitis. In the posterior segment patients may develop vitritis, ischemic retinal vasculitis with neovascularization, occlusion of the central retinal vein, optic nerve affection, macular edema, perforating lesions, “wax drops” exudates and retinal detachment with blindness, which can be detected by slit lamp eye examination and retinangiography.38,39
Skin disorders occur in 20 to 30% of the patients. The most frequent lesion is erythema nodosum, used as a marker of acute sarcoidosis since it usually disappears in 6–8 weeks. The simultaneous appearance of erythema nodosum, fever, joint pain and hilar lymphadenopathy is called Löfgren syndrome. Other skin lesions include subcutaneous nodules, psoriasis like plaques, alopecia, hyperpigmented lesions and leukocytoclastic vasculitis.
Joint involvement is present in 80% of cases. It can be in the form of migratory polyarthritis and/or persistent arthritis, as well as granulomatous tenosynovitis. When polyarthritis, rash and uveitis occur in children under 5 years, it is recognized as Blau syndrome.40
Nervous system involvement occurs in 10% of patients, usually with involvement of II and VII cranial nerves as a result of granulomatous meningitis. However, CNS involvement is found in up to 25% of autopsy cases of sarcoidosis.
Cardiac involvement is diagnosed clinically in less than 5% of patients with sarcoidosis, although it has been found that 20 to 30% of post-mortem studies have lesions in the conduction system, which can cause arrhythmias and sudden death. Echocardiographic findings include ventricular dysfunction and decreased ejection fraction of the left ventricle; however, histological evidence of myocardial biopsies requires cardiac catheterization.
Exocrine glands are frequently affected. Sarcoidosis can involve parotid and minor salivary glands in up to 60% of cases; granulomatous pancreatitis has also been reported.
Renal involvement occurs in 10% of patients due to hypercalcemia, nephrocalcinosis and nephrolithiasis.
Clinical genitourinary involvement has been reported in 0.2% of cases, and in 5% of autopsy studies, the most frequent alteration being scrotal mass.41-48.
The diagnosis of childhood sarcoidosis is performed with the same criteria established for adults, published by the American Thoracic Society and the European Respiratory Society in 1999:
- 1.
Compatible clinical scenario.
- 2.
Histological evidence of noncaseating granulomas in biopsies obtained from the affected organs.
- 3.
Exclusion of other pathological processes that may present with a similar clinical or histopathological presentation, especially mycobacterial and fungal infections and other immunological processes.
There is no definitive test available to confirm the diagnosis of sarcoidosis. However, auxiliary studies are:
- 1.
Cytological analysis of bronchoalveolar lavage of patients with sarcoidosis shows a predominant lymphocyte cellularity in > 90% of cases. Furthermore, a CD4/CD8 ratio > 3.5 by flow cytometry, has sensitivity of 52–59% and specificity of 94–96% for the diagnosis of sarcoidosis.49-51
- 2.
Kveim-Siltzbach test has been used for sarcoidosis diagnosis since 1941. It consists of an intradermal inoculation of a suspension obtained from spleen tissue of patients with sarcoidosis; a skin biopsy should be performed 6 weeks after inoculation in search of granulomas. The test has sensitivity of 75% and specificity > 90%, but currently its clinical application is restricted because of the difficulty in obtaining the reagent; besides, it requires that the patient has not received steroids, and some authors have discouraged it because of the risk of transmission of infectious diseases.51,52
- 3.
Serum angiotensin converting enzyme (ACE) quantification has been used in the diagnosis of the disease, because 80% of pediatric patients with sarcoidosis have elevated ACE. It is also useful as a marker of disease activity as the enzyme levels descend when patients are in remission.
- 4.
Measurement of soluble receptor of interleukin-2 (sIL-2R) is a test that has been very useful in evaluating the activity of the sarcoidosis.53-55
- 5.
Serum and urinary calcium are both useful in the diagnosis and monitoring of disease progression, as hypercalcemia occurs in 20% of cases and hypercalciuria in up to 60% and depend on the activity and the extent of the disease.56-60
We present the case of a twelve-year-old male with no relevant family history, parents and four siblings healthy. He was the product of a first pregnancy, born at term by vaginal delivery without perinatal complications. He had achieved age-appropriate psychomotor development and had a complete immunization schedule. He had no medical history except for chickenpox at the age of 6 years without complications.
Symptomatic disease began one year earlier with his first visit to the hospital, presenting dry cough in isolated bouts. During the last 6 months he had loss 12kg of weight, and had asthenia, hyporexia, paleness, nausea and occasional vomiting of gastric content. Four months earlier, a mass growing in the left supraclavicular region and painless subcutaneous nodules appeared on both forearms. A week before admission he attended a regional hospital because of abdominal colic pain in the right flank and tenderness to lumbar percussion. Abdominal ultrasound showed hepatomegaly and right nephrolithiasis, so he was referred to our institution.
On physical examination, the pacient was conscious and well oriented, emaciated, pale, with equal sized pupils reactive to light, without abnormal findings in ear, nose or throat. Cranial nerves function was preserved. The neck showed no jugular engorgement; he had cervical nodes of 0.5 to 1cm, and a 2cm mobile, painless node was present in the left supraclavicular zone which had no erythema or temperature increase. He had tachypnea and light intercostal retraction, respiratory movements were normal and breath sounds were normal without rales or wheezing. Heart sounds were rhythmic and without murmurs. The abdomen was soft. Hepatic border was 6cm and spleen border 4cm below the costal edge, bowel sounds were normal. He had Tanner II genital development stage and an asymmetric scrotum. Left testicle had a painless increase of volume with no transillumination. Extremities were hypotrophic with erythema nodosum located in forearms and legs, had symmetrical peripheral pulses, normal tendon reflexes with no pyramidal signs.
Initial laboratory test results were: hemoglobin 14.4g/dl, leukocytes 7,600/mm3, neutrophils 48%, band forms 4%, lymphocytes 37%, monocytes 7% platelets 286,000/mm3, erythrocyte sedimentation rate 38mm/h. C-reactive protein <0.319mg/dl. Blood chemistry showed glucose 91mg/dl, creatinine 1.5mg/dl, BUN 17mg/dl, uric acid 6mg/dl, serum proteins 8.6g/dl, albumin 3.2g/dl, globulins 5.4g/dl, total bilirubin 0.36mg/dl, direct bilirubin 0.9mg/dl, indirect bilirubin 0.27mg/dl, ALT 35U, AST 26U, cholesterol 120mg/dl, triglycerides 160mg/dl, Na 129mmol/l, K 3.6mmol/l, Cl 98mmol/l, Ca 13mg/dl, P 4.02mg/dl, Mg 2.4mg/dl, serum osmolality 268mOsm/l. ABG without oxygen: pH 7.43, pCO2 32.4mmHg, pO249.9mmHg, HCO3 21.3mmol/l, SO2 83%, lactate 2.4mmol/l, anion Gap 13.6mmol/l, TP 12.4”, TTP 29.3”.
Serologic studies for hepatitis A, B and C, CMV, EBV, varicella zoster, and type I and II herpes simplex virus were negative. ELISA for HIV was negative. Anti-ds-DNA antibodies, antinuclear antibodies (ANA), anti-neutrophil-cytoplasmic antibodies (p-ANCA and c-ANCA) and VDRL were negative.
The urinalysis reported a density of 1.020, pH 6.0, abundant erythrocytes, leukocytes 1–2/field, negative nitrites, protein 75mg/dl, sediment with calcium oxalate crystals and amorphous phosphate.
Twenty four-hour urine collection showed a creatinine clearance of 70.4mg/ml/min, calciuria of 10.9mg/kg/day, phosphaturia of 63.1mg/kg/day. Tubular phosphate reabsorption was 63.3%, phosphorus/creatinine ratio (P/cr) 0.80mg/mg and calcium/creatinine ratio (Ca/cr) was 0.52mg/mg. Twenty four-hour oxaluria was 53.7mg (<38mg) and 24-h citric acid urine excretion, 427mg (100–1300mg).
Serum cystatin C, 2.18mg/l (0.53–0.95mg/l), parathyroid hormone (PTH), <1.20pg/ml (10–55pg/ml), calcitriol (1, 25-dihydroxy-vitamin D), 30.7pg/ml (19.9–79.3pg/ml), angiotensin converting enzyme (ACE), 272U/l (13–100U/l). Alpha-fetoprotein, 1.07ng/ml (<10ng/ml), human chorionic gonadotropin, 1.20mIU/ml (<5mIU/l).
PPD test with candidin control was negative. BAAR search in three samples of gastric juice was negative.
Bone marrow aspirate showed decreased cellularity, diminished megakaryocytes, metamyelocytes 2.5%, band forms 10.5%, mature neutrophils 35%, eosinophils 3%, lymphocytes 23.5%, blasts 1.5%, and normoblasts 24%.
Serum immunoglobulins: IgA 724mg/dl (108–325mg/dl), IgE 82.6mg/dl (0–100mg/dl), IgM 175mg/dl (70–150mg/dl), IgG 2710mg/dl (770–1510mg/dl).
Complement: C3 113mg/dl (90–180mg/dl), C4 16.6mg/dl (10–40mg/dl).
Nitroblue tetrazolium slide test and chemiluminescence at rest and after stimulus were normal. Soluble Interleukin-2 receptor (sIL-2R), 3230U/ml (406 −1100U/ml).
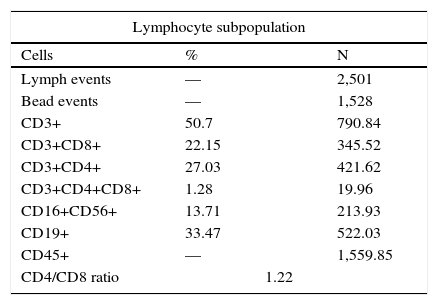
Flow cytometry showed a normal quantity of T and B lymphocytes with a CD4/CD8 ratio of 1.22 (Table 2).
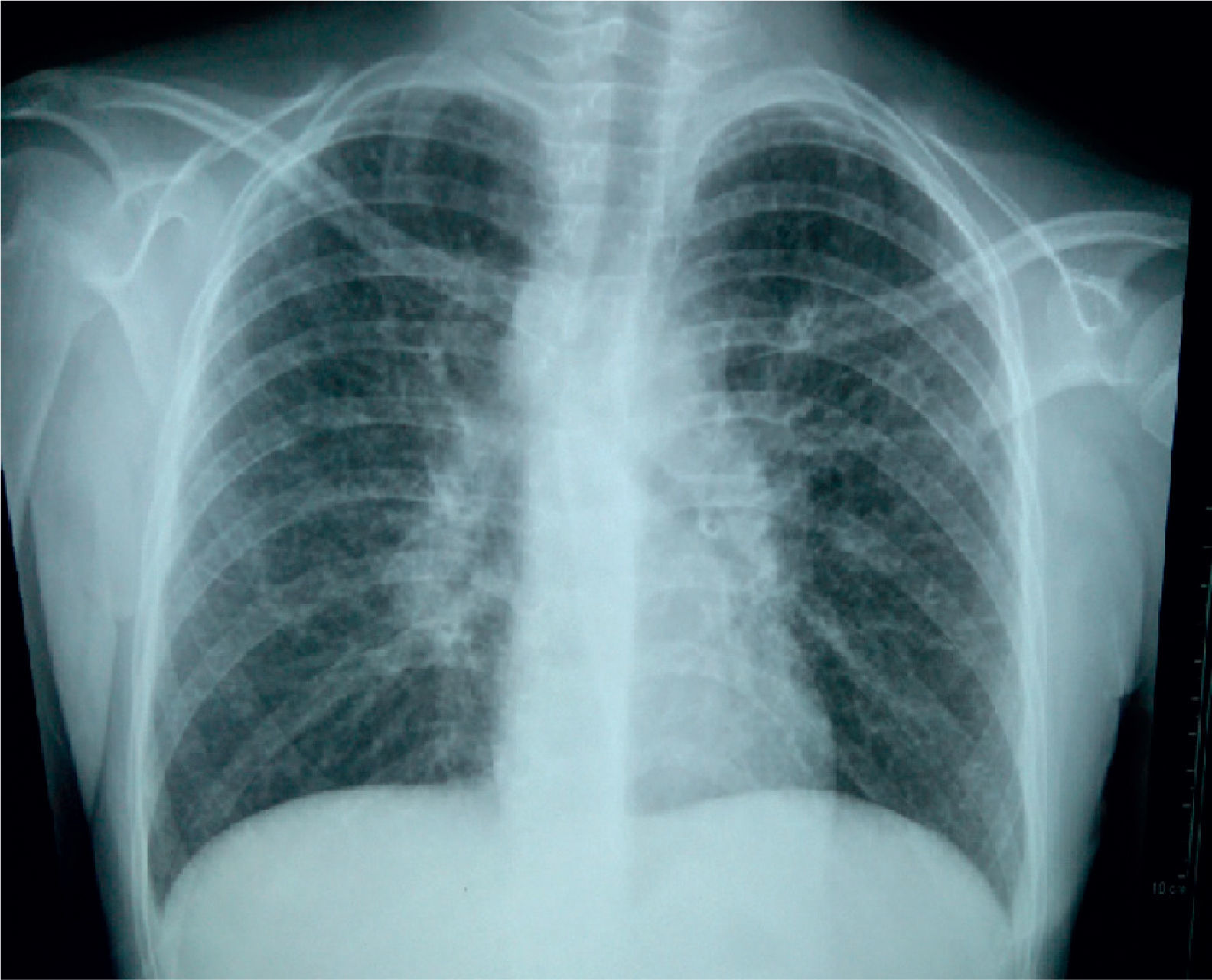
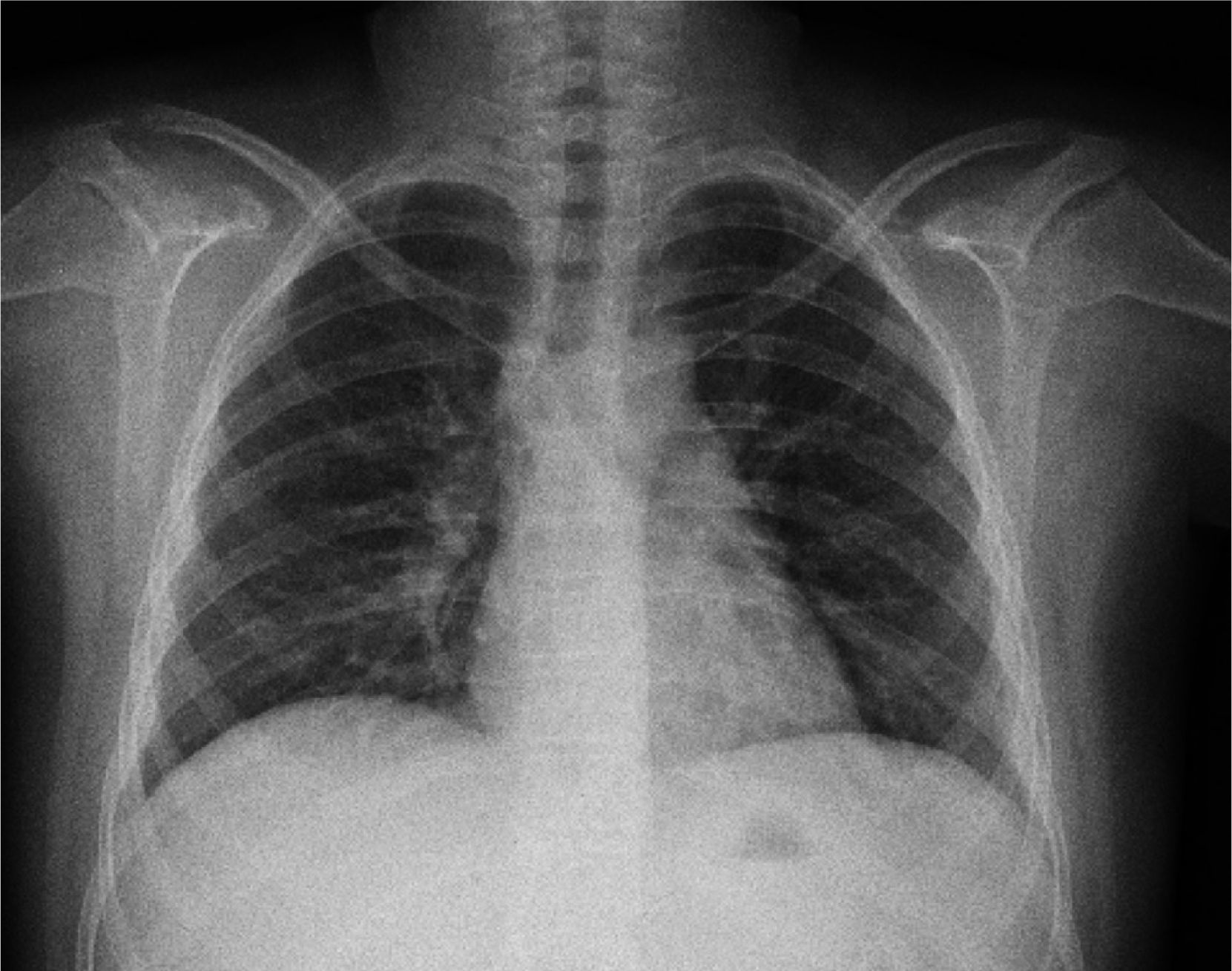
The initial chest X-ray showed diffuse reticular and micronodular infiltrates with bilateral hilar lymphadenopathy (Figure 1).

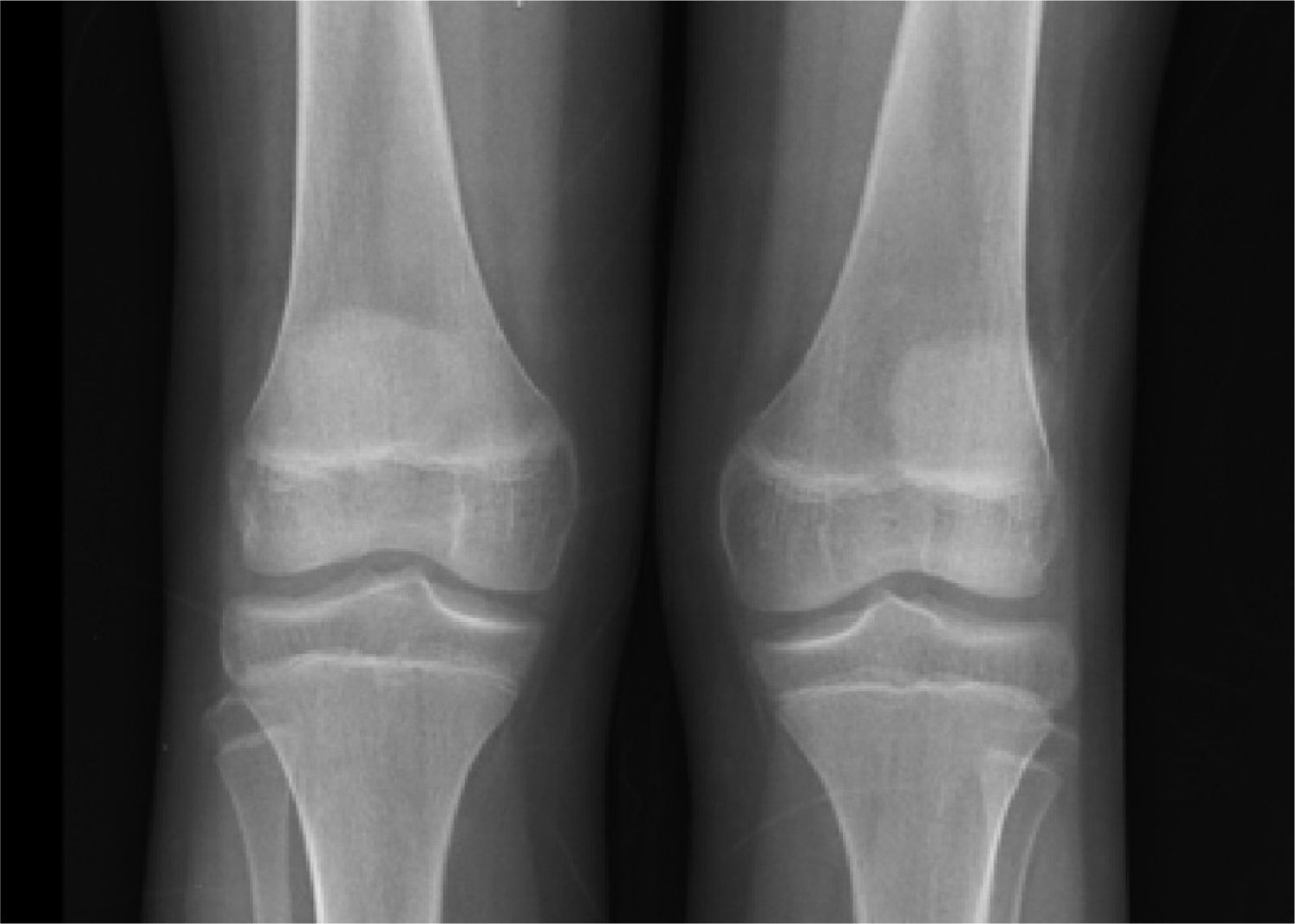
X-rays showed normal skull, and long bones with evidence of decalcification (Figure 2).

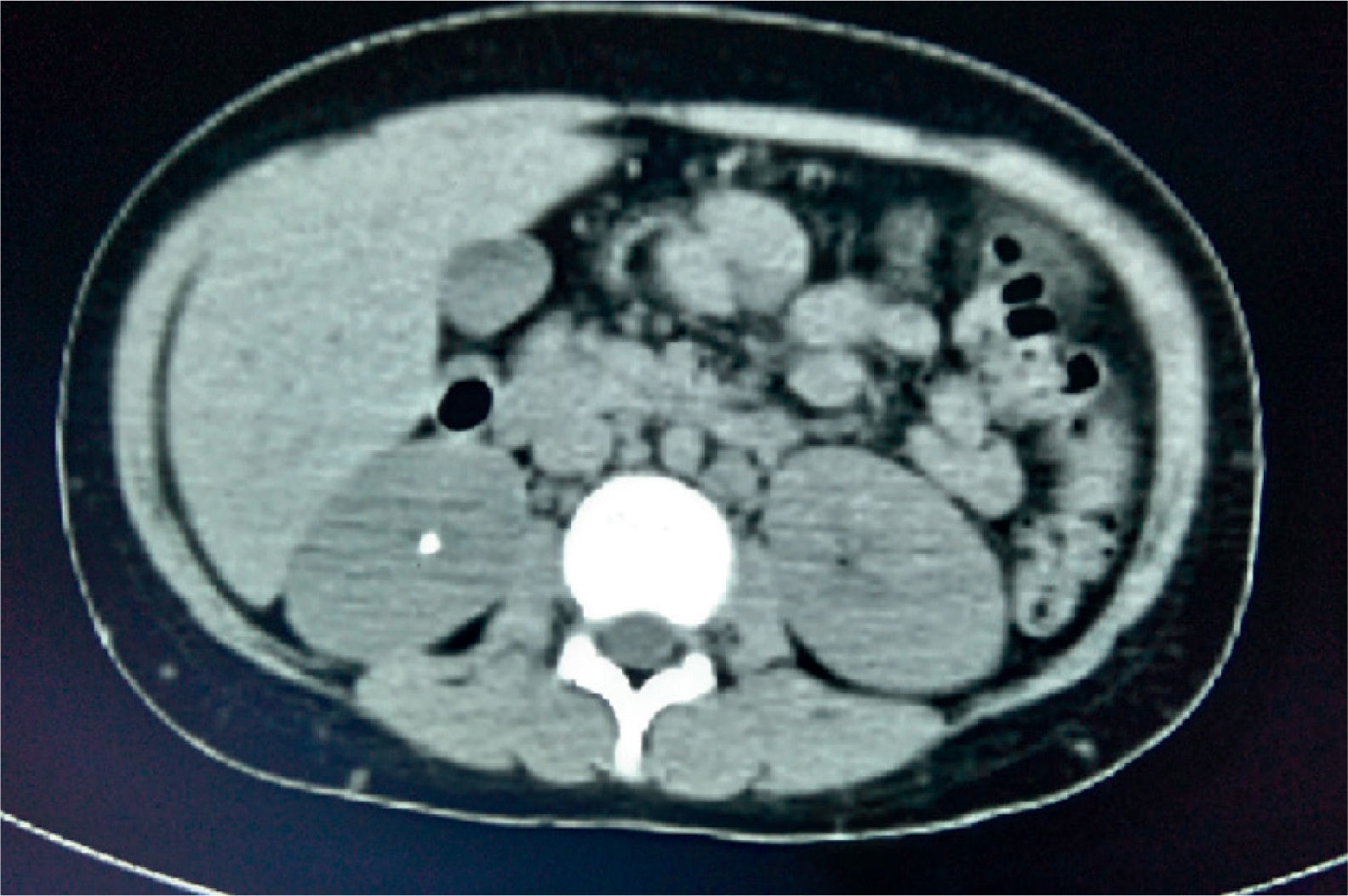
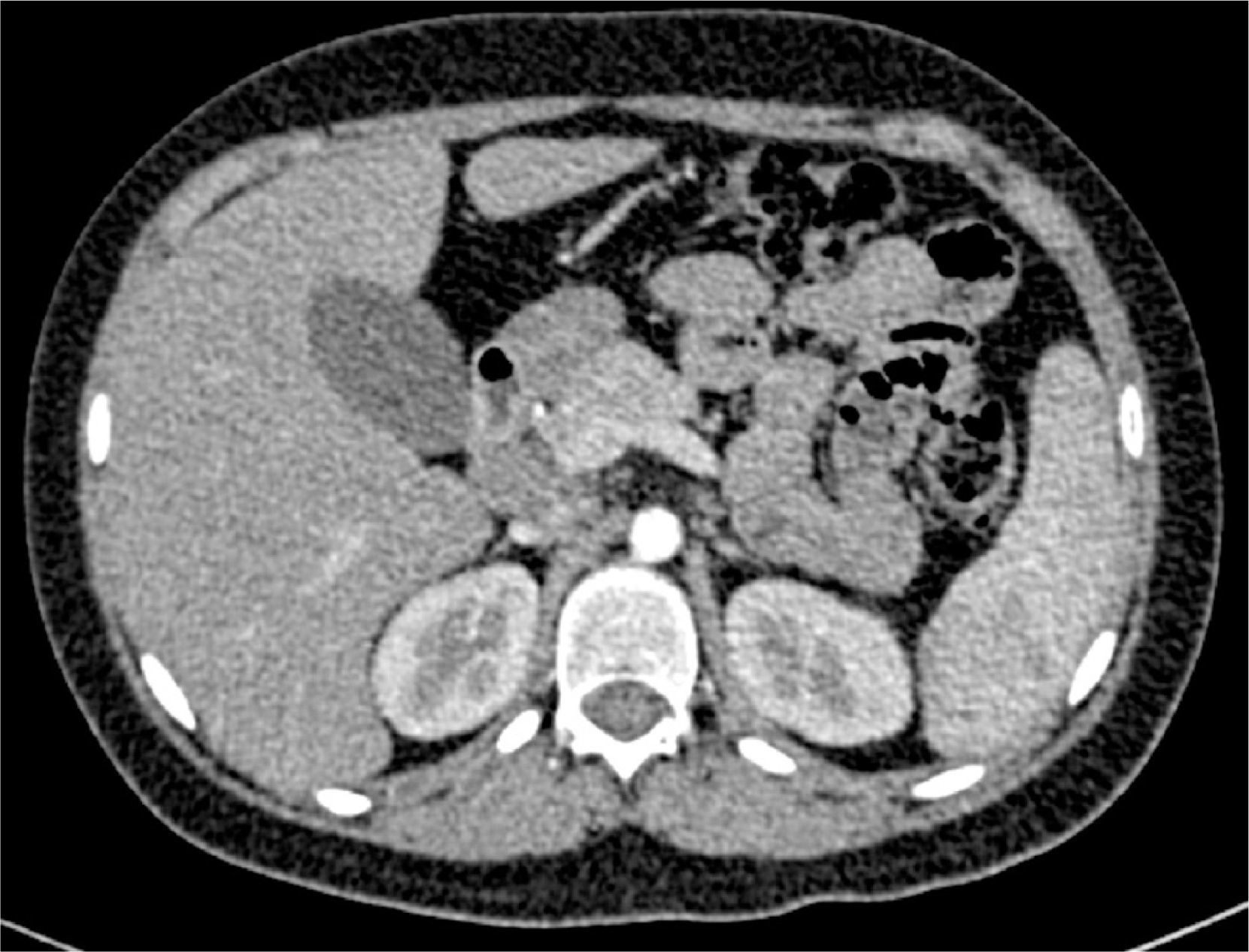
Thoracoabdominal computed tomography showed lymph node clusters in the mediastinum, mesentery, retroperitoneal and inguinal regions, bilateral diffuse micronodular infiltration of the lungs, hepatomegaly, splenomegaly, and right renal calculi (Figure 3).

Renal ultrasound reported normal sized kidneys with regular morphology, appropriate corticomedullary relationship with no dilatation of collecting systems. The right kidney had a stone of 6.7mm located at the sinus. Doppler perfusion image was normal.
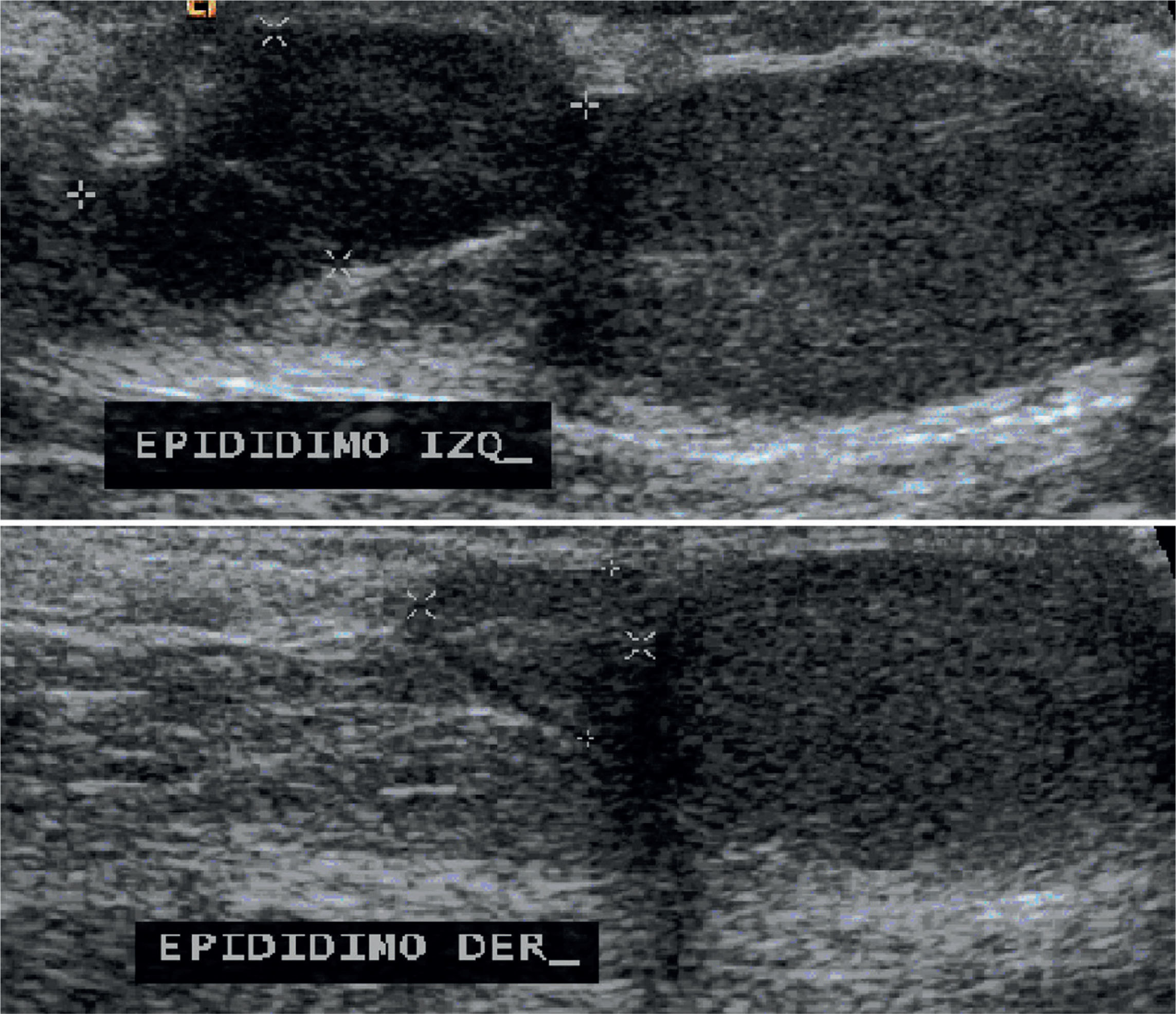
Testicular ultrasound showed both testes and right epididymis of normal size, whereas the left epididymis was enlarged, lobed and with well-defined borders, a size of 15×7×15mm and central vascularity on Doppler mode (Figure 4).

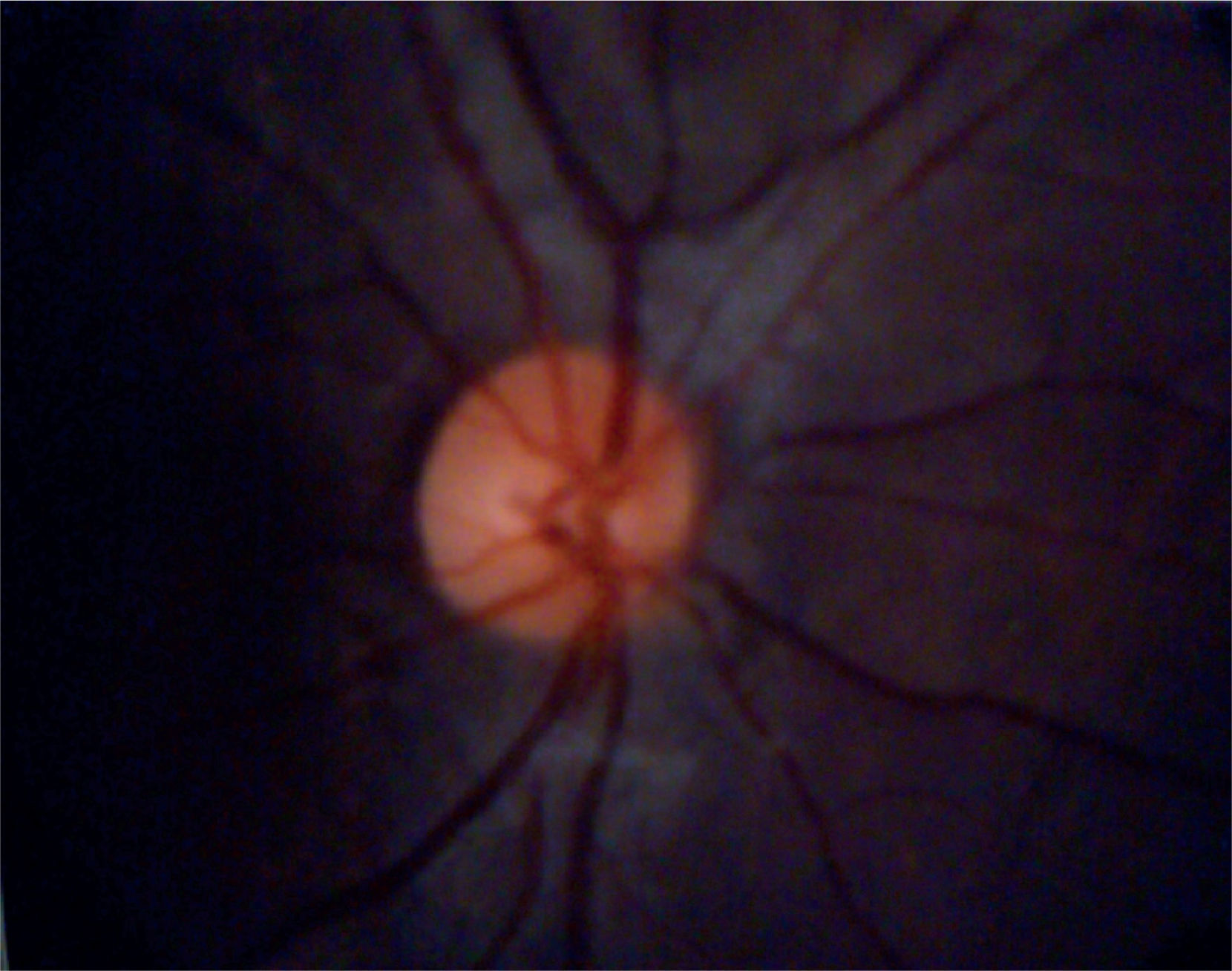
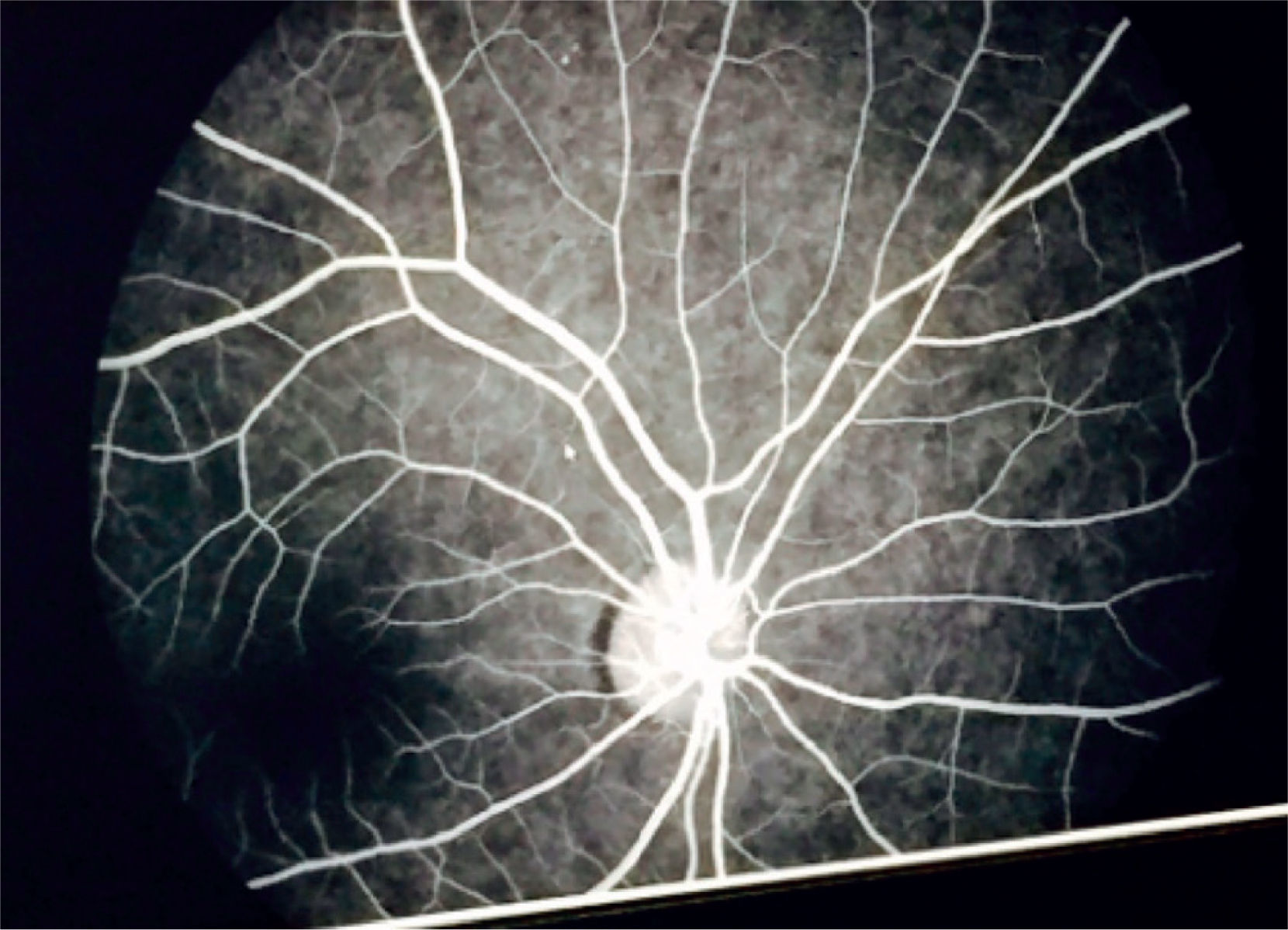
Eye fundoscopy showed temporal and inferior-nasal periphlebitis which are vascular changes characteristic of sarcoidosis (Figure 5). However, fluorescein angiography reported a 3/10 excavation in both eyes, central emergency of vessels, attached retinas, and normal choroidal phase without alterations in vascular pathways (Figure 6).


Echocardiogram showed an anatomically normal heart, left ventricle ejection fraction 72%, shortening fraction 40%, right ventricle systolic pressure 17mmHg, diastolic interventricular septum 9mm.
Pulmonary function test results (quality grade A, met ATS acceptability and repeatability criteria). Spirometry: FEV1 / FVC 89 (102), FVC 2.73 (91%), FEV1 2.43 (93%). Plethysmography: SVC 2.72 (90), IC 1.85 (74), ERV 0.87 (164), TGV 1.27 (71), RV 0.40 (45), TLC 3.11 (73) RV/TLC 13 (57), compatible with a mild restrictive pattern.
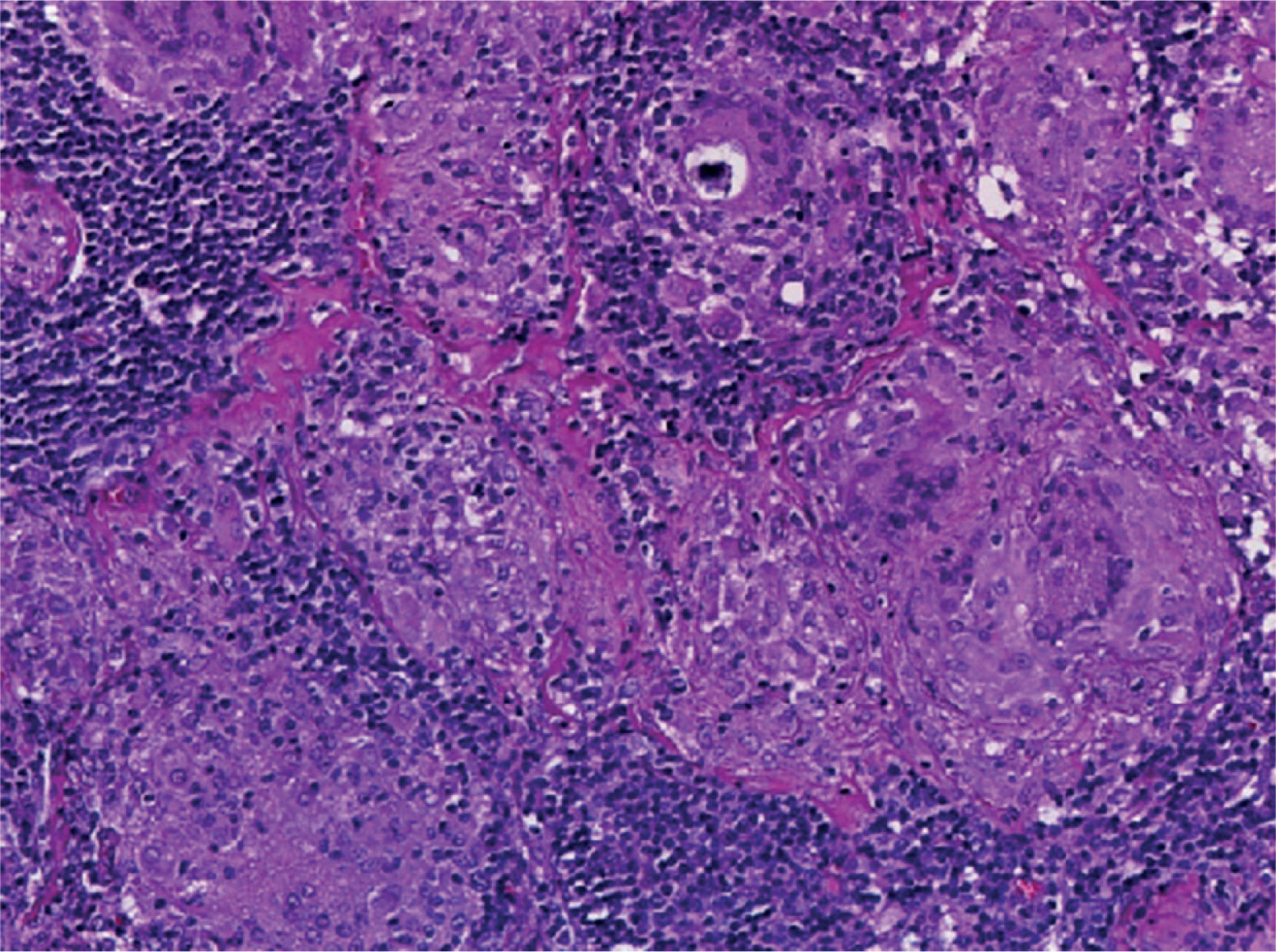
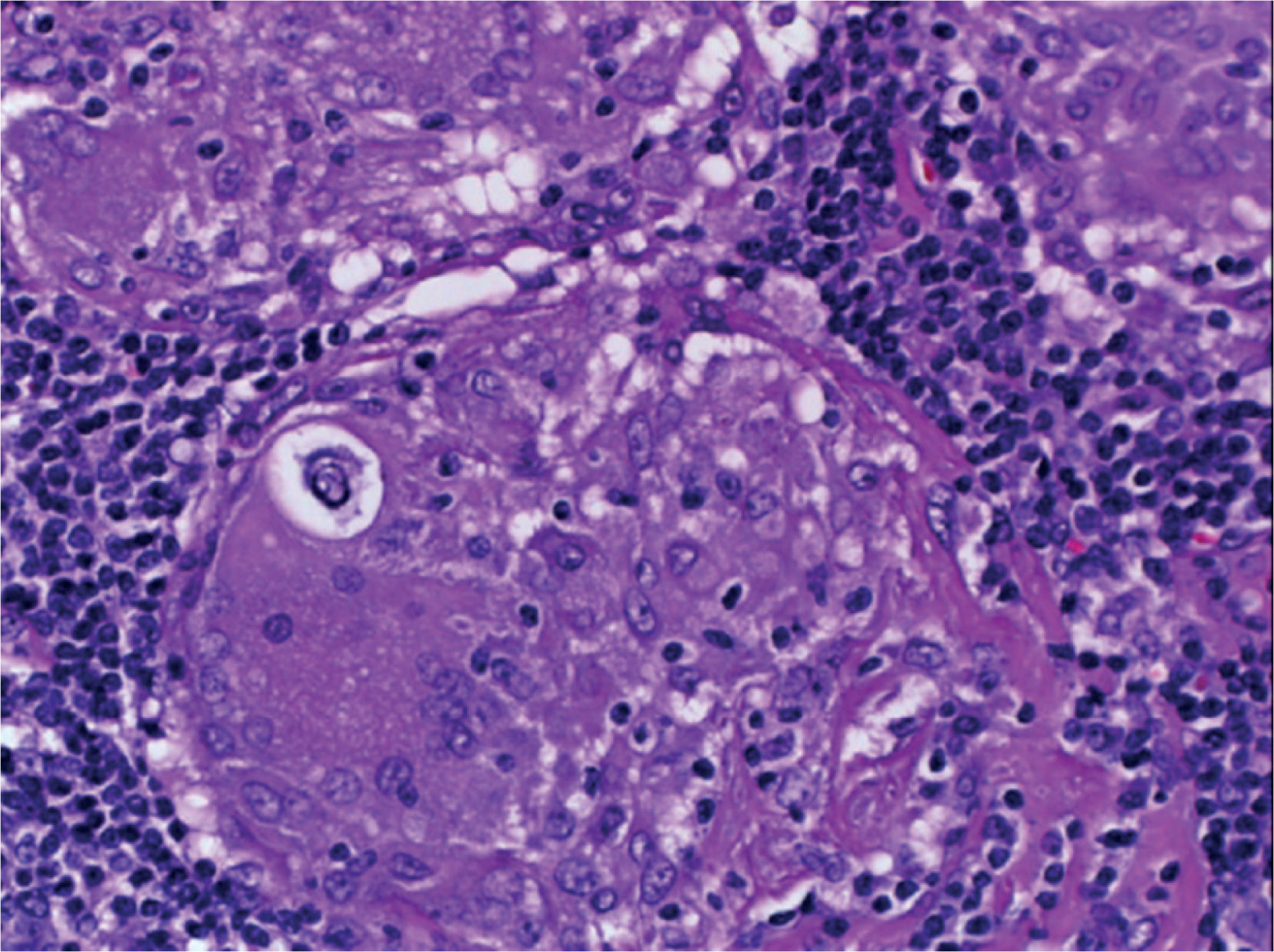
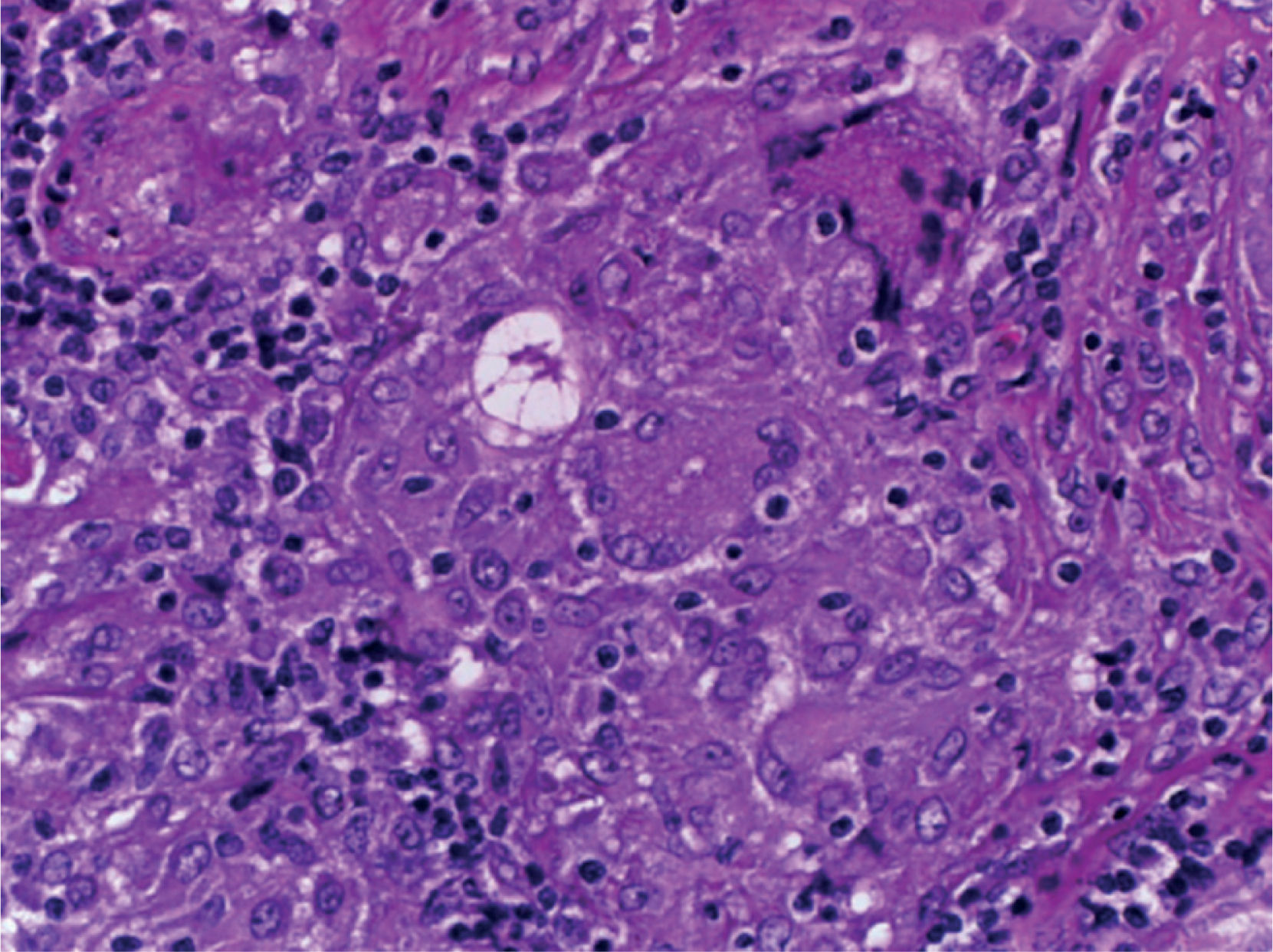
Supraclavicular lymph node biopsy. Hematoxylin-eosin, Grocott, Ziehl-Neelsen and PAS staining were performed. Histological sections showed loss of lymph node architecture, secondary to the formation of multiple confluent noncaseating granulomas (Figure 7), made of numerous epithelioid macrophages, giant Langhans cells and other cells of foreign body-type. Concentric calcifications or Schaumann bodies were observed occasionally (Figure 8) as well as asteroid bodies (Figure 9). Neither special stains nor PCR nor cultures identified Mycobacterium tuberculosis. Histopathological diagnosis: supraclavicular lymph nodes with chronic noncaseating granulomatous lymphadenitis compatible with sarcoidosis.



In the early days of hospital stay, the patient had headache and Kernig and Brudzinski signs. Cranial computed tomography showed adequate differentiation of gray and white matter, normal ventricular system and vascular structures without meningeal alterations. Cerebrospinal fluid (CSF) was transparent, proteins 30mg/dl, glucose 49mg/dl, leukocytes 1/field. Gram stain and cultures were negative. PCR test for Mycobacterium tuberculosis in CSF was negative.
Treatment was initiated with prednisone 2mg/kg/day and hydrochlorothiazide 1.2mg/kg/day. The patient showed clinical improvement in his general condition, remission of hepatomegaly and splenomegaly, erythema nodosum, as well as disappearance of lymphadenopathy and the testicular mass. Mediastinal lymph nodes and pulmonary infiltrates also disappeared (Figure 10).

Four months later, the patient was asymptomatic, had recovered 15kg of weight and had resumed school. In follow up, abdominal tomography showed remission of retroperitoneal lymphadenopathy and no evidence of renal calculi; spirometry and plethysmography had a normal pattern. Laboratory tests showed serum calcium 9.0mg/dl, phosphorus 5.8mg/dl, creatinine 0.8mg/dl, uric acid 6.2mg/dl, 24-hour calciuria 0.93mg/kg/day, and ACE was 71U/l. According to these values, it was considered that the sarcoidosis was in remission.
3DiscussionThis case is about an adolescent male with late-onset pediatric sarcoidosis who had serious affection of the general state leading to cachexia. He also had lung damage, hepatomegaly, splenomegaly, adenopathy, erythema nodosum and a scrotal mass. The diagnostic approach to patients with hepatosplenomegaly, allowed to rule out several infectious diseases such as tuberculosis, systemic mycosis, syphilis, Epstein Barr virus (EBV) and human immunodeficiency virus (HIV), as well as infiltrative disorders, such as leukemias, lymphomas, metastatic tumors and hemophagocytic lymphohistiocytosis by computed tomography, and bone marrow and lymph node biopsies. Systemic vasculitis and other autoimmune diseases were discarded as well as testicular tumors based on the findings of testicular ultrasound and normal levels of the tumor markers alpha-fetoprotein and beta-gonadotrophin.61
The diagnosis of sarcoidosis was suspected because of the presence of a systemic disease with hypercalcemia, hypercalciuria and urolithiasis, accompanied by elevated levels of ACE and sIL-2R. It was confirmed by histopathological study of a lymph node which showed the presence of multiple noncaseating epithelioid granulomas formed by macrophages, giant Langhans cells, asteroid bodies and Schaumann bodies. Cytological analysis of bronchoalveolar lavage and the Kveim-Siltzbach test could not be performed.
On admission, the patient had chronic respiratory failure type I (hypoxemia, normocarbia and hyperlactatemia) with no cardiovascular repercussion. Chest radiography showed diffuse reticular and micronodular infiltrates with bilateral hilar lymphadenopathy concordant with the stage II of the American Thoracic Society for thoracic sarcoidosis classification. Likewise, spirometry and plethysmography reported a decrease in FEV1/FVC, compatible with mild restrictive lung disease as described in the first two stages of pulmonary sarcoidosis, and which normalized after prednisone treatment.
During his hospital stay, the patient also presented headache and meningeal signs which we attributed to sarcoidosis infiltration of the meninges, since CSF cytochemical and microbiological studies were normal as well as cranial computed tomography. These clinical findings also disappeared after prednisone administration.62,63
Although the patient never had ocular manifestations, ophthalmological examination was carried out due to the high frequency of ocular involvement in sarcoidosis. Periphlebitis was compatible with uveal sarcoidosis. As previously described, this is the treatment of choice since sarcoidosis eye injuries usually improve rapidly after prednisone administration, preventing disease progression and the appearance of other irreversible damage which may lead to blindness.64
Hypercalcemia of sarcoidosis traditionally has been attributed to calcitriol production by activated macrophages and monocytes from granulomas, which in turn increase the intestinal absorption of calcium and osteoclast activity with bone resorption and increased levels of serum calcium. However, in this case, the determination of calcitriol (1, 25-dihydroxy-vitamin D3) was found to be within the reference interval, and suppressed PTH levels; this is indicative of inhibition of the parathyroid glands due to elevated serum calcium and, on the other hand, hypercalcemia was probably caused by another autonomous factor such as PTHrP production by activated macrophages, which could not be measured in this case.65
Initially the patient had acute renal failure, but no renal replacement therapy was needed. Hypercalcemic hypercalciuria with decreased tubular phosphate reabsorption, which was expressed as calcium oxalate and calcium phosphate, caused him kidney stones. While the acute colic pain is uncommon in children with urolithiasis, this patient presented right flank pain accompanied by microscopic hematuria. As part of the medical management, treatment with hydrochlorothiazide was provided in order to reduce the renal calcium excretion and decrease the formation of calcium oxalate and calcium phosphate calculi, achieving remission of hypercalciuria and expulsion of renal calculi (Figure 11).66–72

In this patient no studies of histocompatibility antigens (HLA) were performed. Regarding immune function, moderate polyclonal hypergammaglobulinemia was observed, with appropriate levels of C3 and C4, as described in patients with sarcoidosis. The phagocytic function tests nitroblue tetrazolium slide test and chemiluminescence at rest and after stimulus were normal, so the possibility of chronic granulomatous disease was discarded.
While flow cytometry on peripheral blood reported a total number of lymphocytes within the normal interval, the identification of the major lymphocyte subpopulations showed 50.7% of T cells, 33.4% of B lymphocytes and 13.7% of NK cells with one CD4/CD8 ratio = 1.22. This could explain the immune paradox observed in skin tests because it was considered that the patient had normal cellular and humoral immunity.73-77
Finally, it should be noted that childhood sarcoidosis is an underdiagnosed disease in Mexico, so there is no clinical or epidemiological information about this condition in our population. This clinical case may contribute to the knowledge of sarcoidosis in children whose clinical symptoms are nonspecific. In the few reported cases, sarcoidosis has been misdiagnosed as juvenile idiopathic arthritis, tuberculosis or systemic fungal infections. However, the involvement of multiple organs may lead to serious disability and/or death, highlighting the importance to consider sarcoidosis in the diagnostic approach of patients with generalized lymphadenopathy, splenomegaly and hepatomegaly in order to initiate glucocorticoid therapy, which is the most effective treatment to prevent disease and stop its progression in most of these patients.
Ethical disclosureProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of interestThe authors declare no conflict of interest of any nature.
Please cite this article as: Zamora-Chávez A, et al. Sarcoidosis en la infancia. Una rara enfermedad sistémica. Bol Med Hosp Infant Mex. 2016;73:117-28.