


Multiple hereditary osteochondromatosis is characterized by the growth of benign cartilaginous tumors in the form of exostosis, mainly in the metaphysis of long bones. A prevalence of 1/50,000 individuals has been documented for this disease.
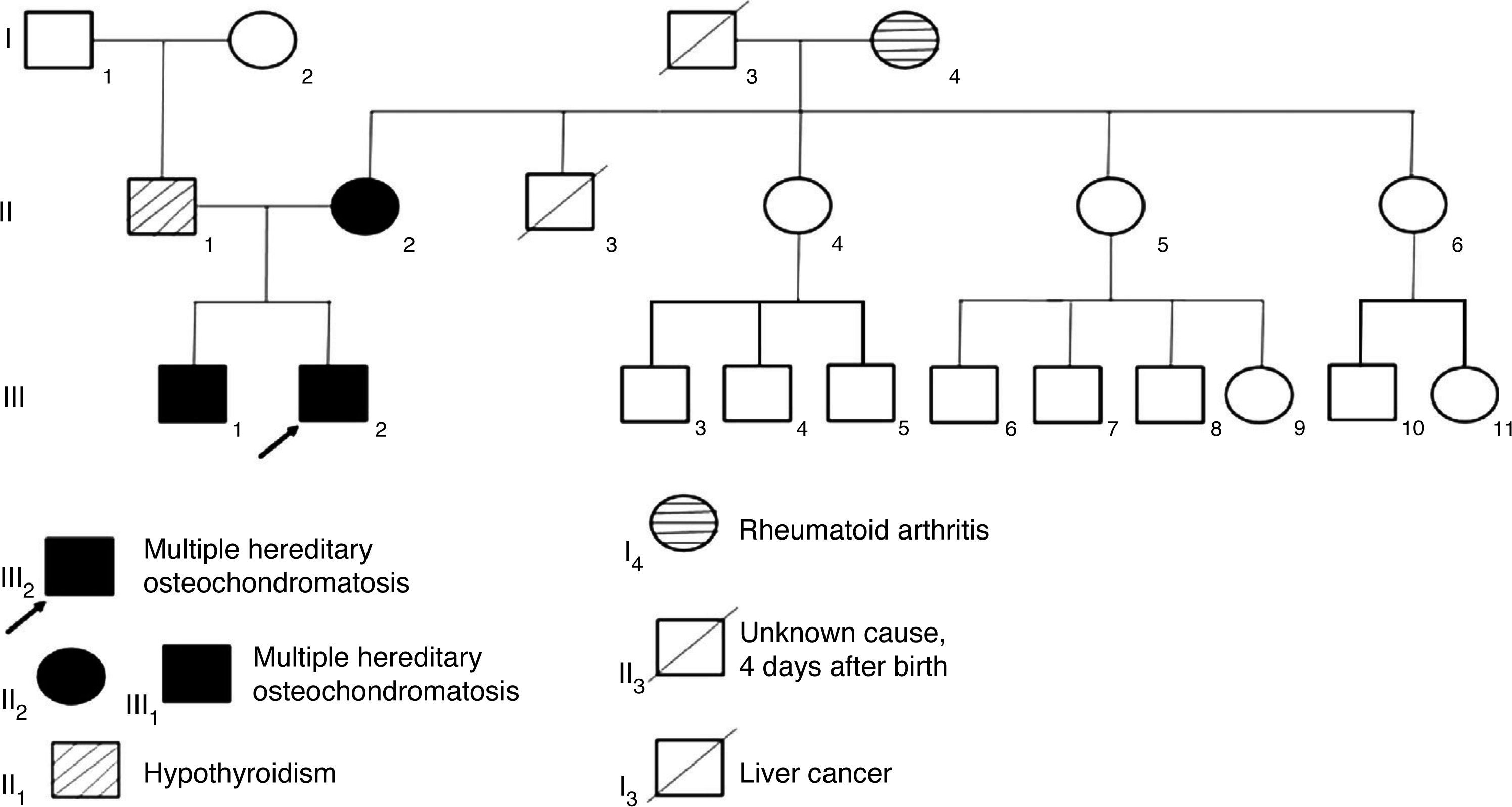
Case reportsThis article presents the clinical information and the autosomal dominant inheritance pattern where exostosin genes (EXT gene family) were affected in three members of a family with multiple hereditary osteochondromatosis. The three patients showed limitation in the range of motion of wrists, shoulders and ankles. Clinical diagnosis was confirmed with radiology and malignancy was ruled out in all patients.
ConclusionsThis disease requires frequent medical evaluation, surgical bone correction when the function is affected, surveillance for malignant transformation, and genetic counseling.
La osteocondromatosis múltiple hereditaria se caracteriza por el crecimiento de múltiples tumores benignos, cartilaginosos, que crecen en forma de exostosis predominantemente en las metáfisis de los huesos largos. Se ha descrito una prevalencia de 1/50,000 individuos.
Casos clínicosSe presenta la información clínica y patrón hereditario autosómico dominante, en el que están afectados los genes de exostosina (familia de genes EXT) en tres miembros de una familia con osteocondromatosis múltiple. Los tres pacientes han presentado alteraciones en los arcos de movimiento de muñecas, hombros o tobillo. El diagnóstico clínico fue confirmado con estudios radiológicos y no hay evidencia de que las lesiones se hayan malignizado.
ConclusionesEsta entidad requiere de supervisión periódica, corrección quirúrgica de las deformaciones que limiten la función, vigilancia de la transformación maligna y consejería genética.
Multiple hereditary osteochondromatosis (MHO) has several synonyms: multiple cartilaginous exostoses, hereditary multiple exostosis, congenital osteochondromatosis, familial osteochondromatosis, hereditary osteochondromatosis, hereditary deforming chondrodysplasia and diaphysial aclasis.1 MHO is an autosomal dominant condition characterized by the growth of multiple benign tumors called osteochondromas. These osteochondromas are cartilage-capped bone tumors growing outward from the metaphysis of long bones, and their number may range from two to hundreds.2 MHO is caused by a defect in metaphyseal osteoclast activity during the remodeling process in childhood and early adolescence. During skeletal development, ostechondromas enlarge and gradually ossify. They stop growing with skeletal maturity after which no new osteochondroma development is observed.1 Tumors are metaphyseal bony protuberances coated by cartilage.
MHO was first described in 1839 by John Hunter in his book “Lectures on the Principles of Surgery”3 and reported prevalence is 1/50,000.4–6 Nowadays, exostosin genes (EXT gene family) are known to express glycoproteins involved in chondrocyte transportation, proliferation and differentiation.7 The gene EXT1 is located in the chromosome 8q24, where 44–66% of mutations responsible of MHO occur. The gene EXT2 is located in the chromosome 11p11-p12. This gene may be altered by up to 30% mutations. The gene EXT3 is located in the chromosome 19p and mutations in this gene may be the cause of some cases of MHO.8
The mean age for diagnosis of MHO is three years and 96% of cases are diagnosed before 12 years of age.4 Osteochondromas usually develop during the time of natural bone growth.9 They arise from the displacement of the lateral side of the growth plate in a direction diagonal to the long axis of the bone and away from the adjacent joint; in consequence, they usually are mushroom shaped and size from one to 20cm. The outer layer of the osteochondroma head is made of hyaline benign cartilage of variable thickness and limited by perichondrium.10 Osteochondromas can produce shortening of the limbs, scoliosis, pathological fractures and pseudarthrosis, bilateral coxa valga, progressive widening of the proximal femoral metaphysis, shortening of the ulna with radial deformation and radio-humeral subluxation, valgus deformation of the knees and ankles, and decreased stature due to deformations and angulations.11,12
Osteochondromas are localized mainly in long bones, such as proximal humerus, distal and proximal femur, tibia and fibula; however, they may also affect the spine, pelvis, ribs and scapulas.13,14.
The deformations most commonly identified are bilateral coxa valga, progressive widening of the femoral proximal metaphysis, ulnar shortening with radial deformation, radio-humeral subluxation, knee and ankle valgus and shortened stature due to deformations and bowing. However, the most common presentation is painless increase in volume, or a slow growing tumor.2 Pain and motor deficits are secondary to the mechanical block. The most feared complication is the development of chondrosarcoma, which occurs up to 25% of cases.11
The imaging method of choice for evaluating the patient with osteochondromas is plain radiography. Generally, the film shows an excrescence, either pedunculated or sessile, with well-defined margins arising from the metaphysis and away from the epiphysis.2 Ocasionally, a widening of the metaphysis is observed. Some osteochondromas arising from the surface of the bone contain cortex and spongy bone, and appear to be a continuation of the original bone.15
The aim of this study was to characterize MHO in three members of a family, to describe the observed inheritance pattern, and to provide genetic counseling.
2Clinical casesThree related cases of MHO were diagnosed. The mother had a de novo mutation which she transmitted to her children in an autosomal dominant pattern (Figure 1). The children’s father did not show apparent osteoarticular problems, but had a history of hypothyroidism, and he was on replacement therapy with levothyroxine.

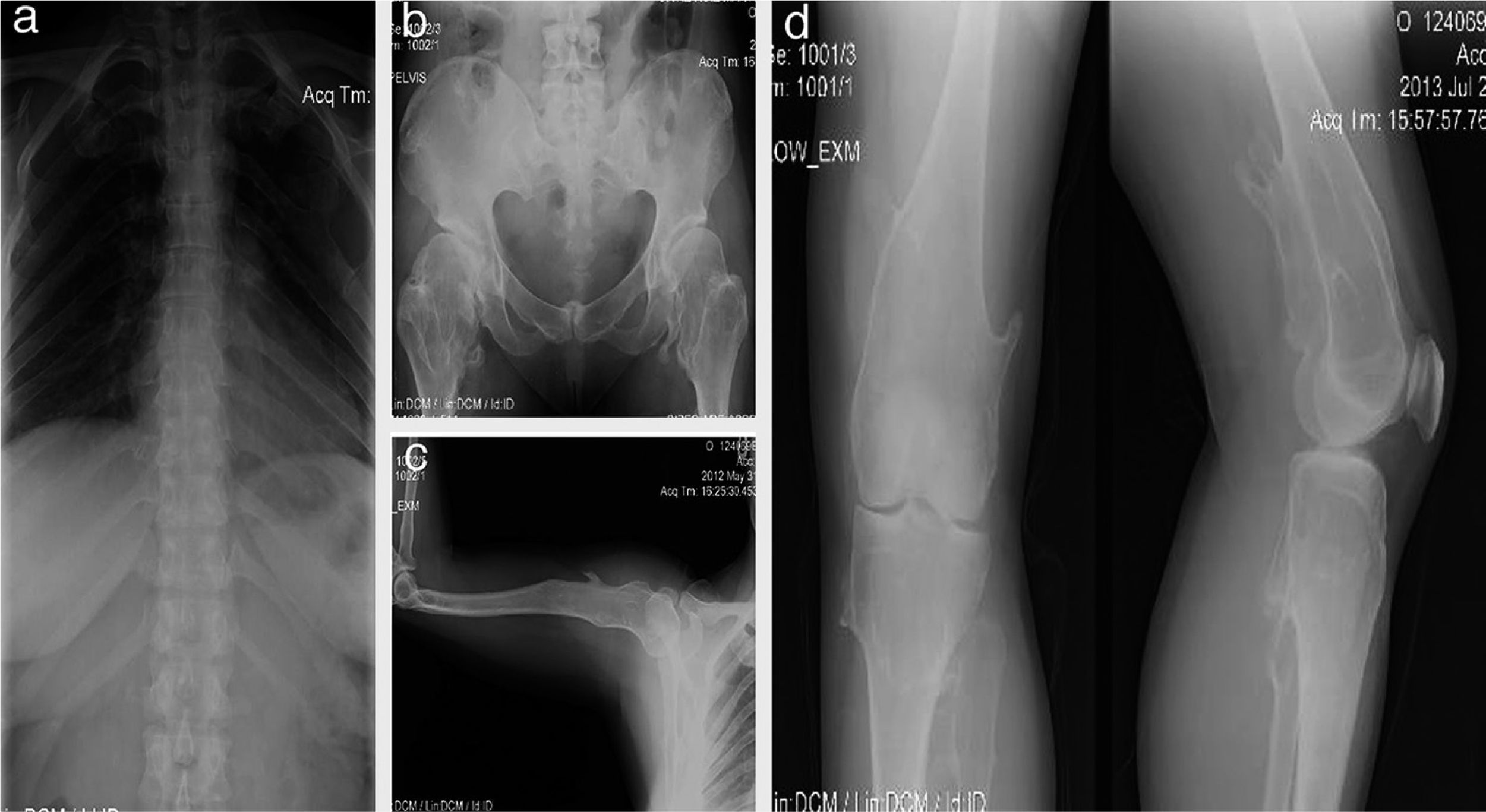
Female patient, 40 years old; since birth, her parents noted articular cracks when they up loaded her although with no apparent pain. At 3 years of age, a disabling lump was detected in the left knee which was painful when walking and required surgery. At age 16, she developed painful deformities in the ankles (in the external malleolus of 4 × 4 × 3cm and in the tibia), so she needed corrective surgery. At age 20, she presented thickening and deformity of the wrists measuring 4 × 5 × 2cm. At the end of bone growth, it was found that the length of the right arm was 2cm greater than the left one (Figure 2).
. A 40-year-old female patient (mother) with exostosis in the first sternocostal joints and in the upper part of the posterior portion of the rib (a), bony protrusions in femoral necks and intertrochanteric zone with bone deformity (b), exostosis in proximal humerus (c) and distal third of posteromedial zone of both femurs and in the posterolateral region of the left tibia (d).")
Case 1 (II2). A 40-year-old female patient (mother) with exostosis in the first sternocostal joints and in the upper part of the posterior portion of the rib (a), bony protrusions in femoral necks and intertrochanteric zone with bone deformity (b), exostosis in proximal humerus (c) and distal third of posteromedial zone of both femurs and in the posterolateral region of the left tibia (d).
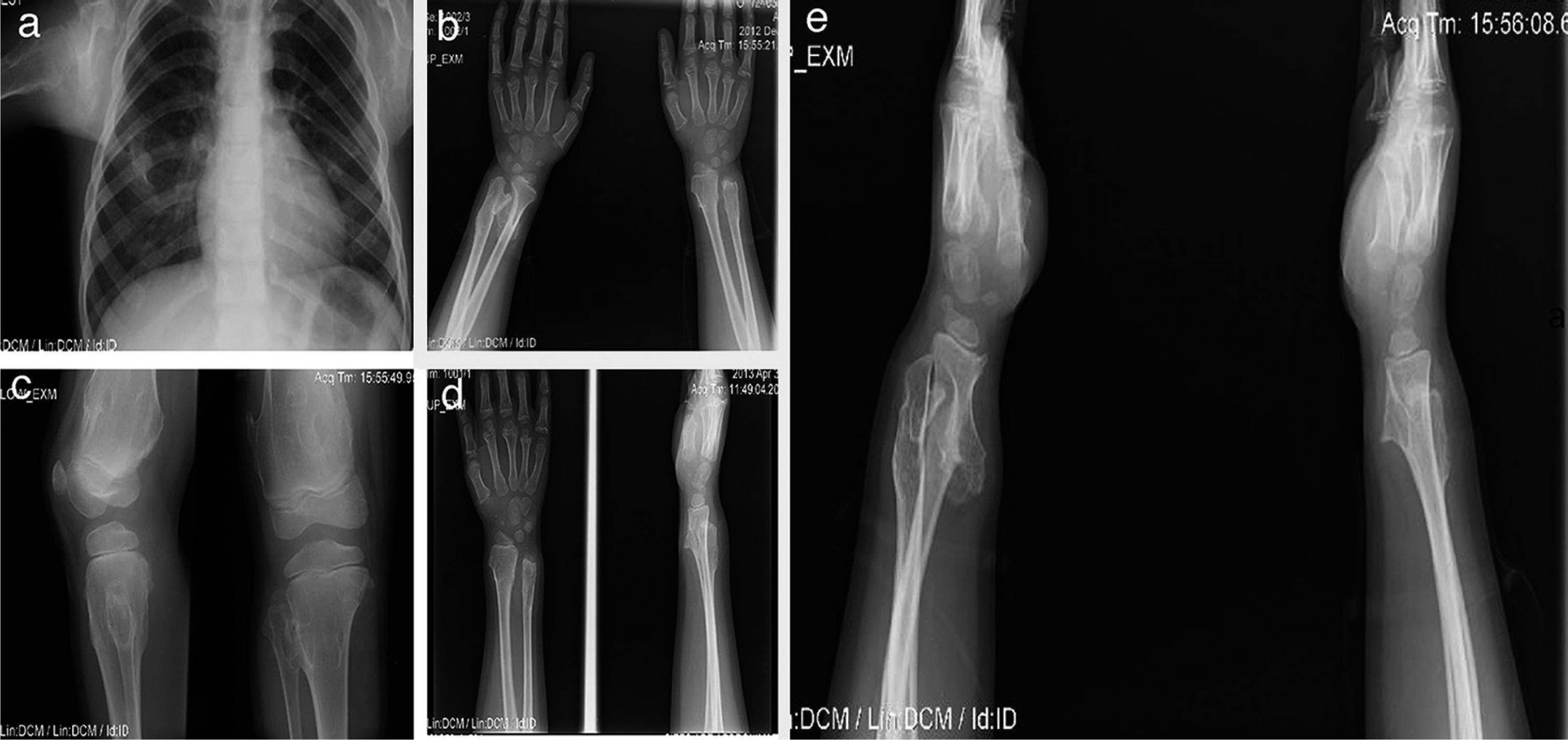
A 12-year-old male patient with a history of subclinical hypothyroidism in the first year of life which did not require treatment. Osteochondromas were noticeable at 2 years of age causing ankle deformities with no fractures. Afterwards, new tumors have aroused in the shoulder, tibia, wrist and femur. He presented limited motion in the wrist and shoulder with occasional pain. His growth has been satisfactory, with height in the 50th percentile and without shortening of the lower limbs (Figure 3).
. A 12-year-old male patient with exostosis in the neck and proximal third of the humerus, in the anterior portion of the fifth rib (a), in the anterolateral zone of both ulnas, suggesting a type III deformity of the Masada classification (b), femur distal third with meta-diaphysial widening (c), and lumps in the anterolateral region of the right tibia and ulna, which increased in size the following year (d, e).")
Case 2 (III1). A 12-year-old male patient with exostosis in the neck and proximal third of the humerus, in the anterior portion of the fifth rib (a), in the anterolateral zone of both ulnas, suggesting a type III deformity of the Masada classification (b), femur distal third with meta-diaphysial widening (c), and lumps in the anterolateral region of the right tibia and ulna, which increased in size the following year (d, e).
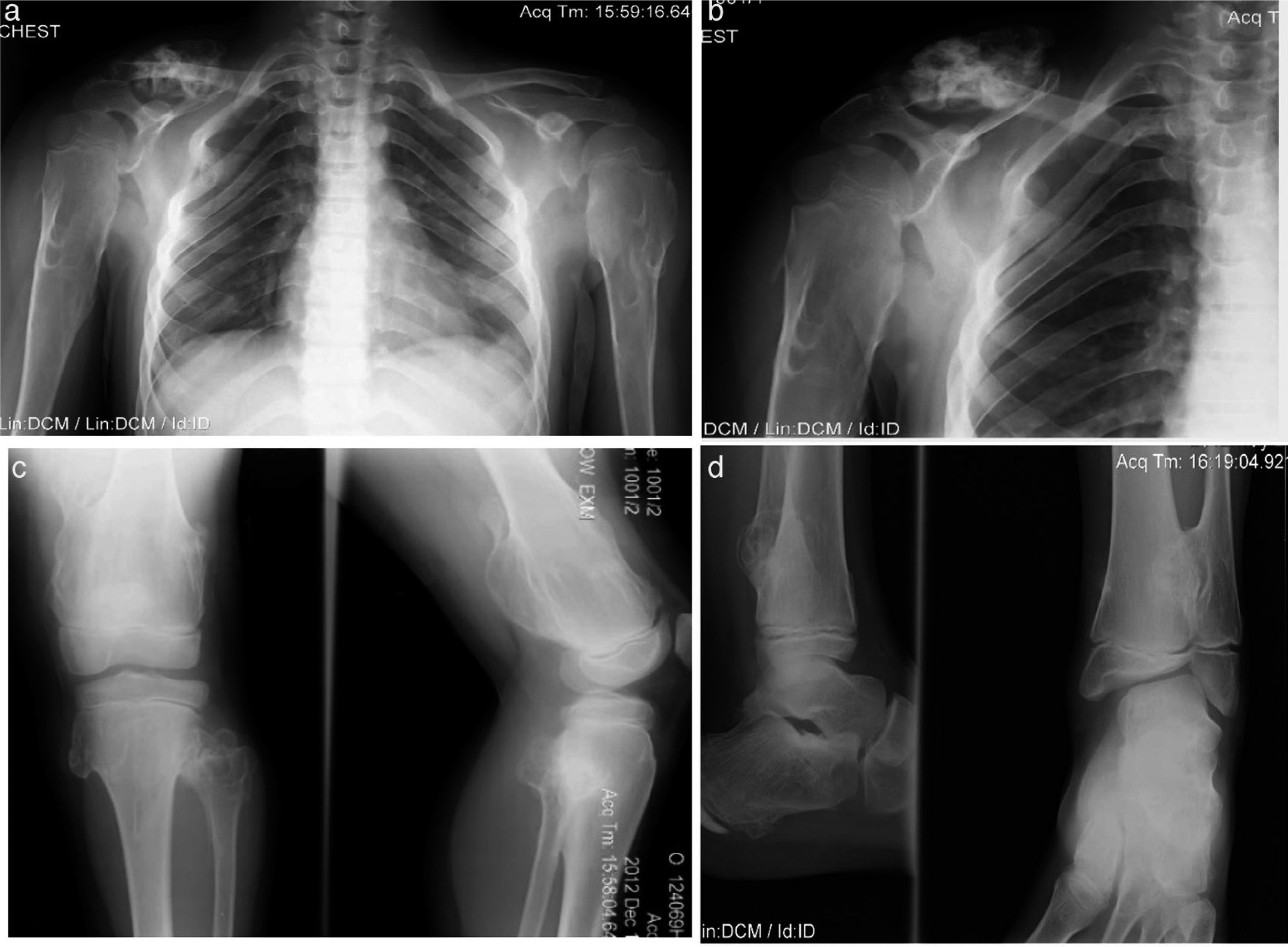
An 8-year-old male patient, diagnosed with subclinical hypothyroidism from 8 months to 2.5 years of age which normalized later. MHO diagnosis was established at 2 years of age because of humeral and wrist tumors. His growth has been satisfactory, without shortening of the lower limbs, as well as weight and height in the 50th percentile. He has limited motion in wrists and shoulders with sporadic pain during certain movements (Figure 4).
. Eight-year old male patient presenting osteocartilaginous lesions with heterogeneous pattern, predominantly radiolucent in right clavicular region (a), sub-metaphysial exostosis in the proximal region of the right humerus (b), distal third of femurs (c), proximal and distal tibia and fibula (c, d). Deformity with medullar widening mainly in humeri and femurs can be observed. Physial regions are not affected.")
Case 3 (III2). Eight-year old male patient presenting osteocartilaginous lesions with heterogeneous pattern, predominantly radiolucent in right clavicular region (a), sub-metaphysial exostosis in the proximal region of the right humerus (b), distal third of femurs (c), proximal and distal tibia and fibula (c, d). Deformity with medullar widening mainly in humeri and femurs can be observed. Physial regions are not affected.
Multiple bone lesions were identified in radiographs, with osteochondromas in metaphysis and epiphysis of long, short and flat bones, of different sizes and shapes, usually with a sessile image of broad base, covered with cortical bone and “mushroom head” shaped cap (IV-V) (Figures 2–4).
3DiscussionThe loss of both alleles of the EXT1 gene has been linked to osteochondromas, suggesting a loss of tumor suppressor activity mechanism.8,16EXTL1, EXTL2 and EXTL3 conform another EXT gene family associated to the MHO, and are considered homologous to EXT1 and EXT2 genes. Heparan sulfate is one of the proteins altered by the lack of processing. In a study of 36 Chinese families it was found that mutations in EXT2 reached 33% whereas in EXT1 reached 14%. The predominant types of mutations were changes of the reading frame and nonsense mutations (80%). These mutations may produce premature termination codons, and hence incomplete non-functional protein. This study shows that the pattern of mutations can vary among different populations.17 According to Wuyts et al., penetrance is 96% among women and 100% in men.1 The affected person has a 50% chance of passing the trait to the offspring.
Regarding differential diagnosis, among the syndromes resulting from the mutation of EXT genes there is the Langer-Giedion syndrome in which there are exostosis, mental retardation, malformations in skull and distal extremities. In Potocki-Shafferse syndrome there are craniofacial abnormalities, mental retardation and epiphyseal exostosis affecting only one side of the body.18 In hemimelic epiphyseal dysplasia, cartilaginous lesions develop in epiphysis causing focal growth of the medial and lateral portions of the epiphysis, with unilateral distribution. This dysplasia most often affects the carpus, tarsus, and lower limbs.19 In contrast, in osteosarcoma, the injuries are usually located in metaphysis of long bones and knees; in radiographs bone erosions are observed in cortical and medullary regions, with poorly defined edges and invasion of soft structures.20
One of the most serious complications of ostoechondromas is their malignant transformation (chondrosarcoma). However, there are not specific guidelines for screening. The warning signs for malignant transformation are growth with irregular margins, presence of a mass of soft tissue attached to the tumor, pain, bone erosion and diffuse calcification with an irregular pattern. Radiological warning signs are radiolucent round nodular lesions or with a pattern in rings.21 In later stages, erosions of the endothelial cortex may be found. None of the cases presented in this study showed suspicious signs of malignant transformation.
Nowadays, the treatment to achieve better effectiveness and better results is surgery. This is reserved for those patients who are symptomatic and/or have deformities caused by osteochondromas. The procedures performed include tumor excision, corrective osteotomy, procedures to align or enlarge bone length, epiphysiodesis and hemiepiphysiodesis.21-24 Of the three members of the family described, II2 was the only case that has required multiple palliative surgeries for pain relief or function restoration (conservation of limb range of motion).
MHO is inherited in an autosomal dominant pattern and is genetically heterogeneous. In about 90% of affected patients, germline mutations of the tumor suppressor genes EXT1 or EXT2 have been found. Diagnosis is established clinically and radiologically. Genetic counseling was offered to the family in order to inform on the risk of recurrence and clinical implications of this disease, to help them make informed medical and personal decisions, and to provide timely and adequate follow-up. Genetic counseling is provided for those families to have information on the nature, inheritance, and the implications of this condition.
Ethical disclosureRight to privacy and informed consentThe authors declare that no patient data appear in this article.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Protection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Conflict of interestsThe authors declare no conflict of interest.
Please cite this article as: Santos-Guzmán J, et al. Osteocondromatosis múltiple hereditaria en una familia. Bol Med Hosp Infant Mex. 2016;73:111-6.