


En México, la microtia presenta una prevalencia de 7.37/10,000 recién nacidos, la cual es más alta que la reportada en otras poblaciones; por ejemplo, en Estados Unidos es de 2-3/10,000 recién nacidos. Se define como la malformación congénita del oído externo caracterizada por un pabellón auricular pequeño y con alteración en su forma. Se observa más frecuentemente de manera unilateral de lado derecho y en varones, y puede presentarse como defecto aislado o asociada con otras alteraciones como atresia y estenosis del conducto auditivo. Representa una de las principales causas de atención en la consulta externa del departamento de genética de instituciones de tercer nivel.
Se considera como una malformación mayor con profundas repercusiones en la función auditiva, y que requiere de una atención multidisciplinaria. En una minoría de casos ha sido posible identificar una causa puramente genética o puramente ambiental, ya que en la mayoría la presentación es multifactorial. Debido a la importancia que representa esta alteración para los diferentes servicios de salud en México, es importante que se conozcan sus bases clínicas, moleculares y hereditarias.
Mexico has a prevalence of microtia of 7.37/10,000 (newborns), 3 times higher than the prevalence reported in other populations (USA 2-3/10,000). Microtia is defined as a congenital malformation of the external ear characterized by a small auricular lobe with an abnormal shape. It is more often unilateral and on the right side. Males are more frequently affected than females. It can occur as an isolated defect or can be associated with other abnormalities such as stenosis of the external auditory canal. In three of the main pediatric hospitals in Mexico, microtia is among the most important causes of attendance in the Genetics Department. Microtia-atresia must be considered as a major malformation with important repercussions in hearing function requiring multidisciplinary medical care in order to limit the disability associated and to provide genetic counseling.
Its etiology is complex. Only in a minor number of cases it has been possible to identify a main genetic component (as in monogenic presentations) or a main environmental cause (as in fetal alcohol syndrome or pregestational diabetes). In most cases this malformation is multifactorial. Due to the relevance that the frequency of microtia atresia has in different health services in Mexico, it is important that all medical professionals are aware of its clinical, molecular and inherited characteristics.
1. Introduction
Microtia (OMIM 600674, OMIM 251800)a is defined as a malformation of the external ear characterized by a small ear and with alteration in its shape. This malformation encompasses a wide spectrum of clinical abnormalities of the ear, which differ with regard to its severity, from minor anomalies to the complete absence of the ear or anotia.1 Its presentation is a public health problem because of its high prevalence and the psychosocial sequelae for the patients.
2. Prevalence
Population studies in some European countries and in the U.S. show a prevalence between 0.83 and 4.34/10,000 births.2-6 Other studies in the U.S. have reported consistent ethnic variations, with a greater prevalence among individuals of Japanese origin (3:1) of the Pacific Islands and in the population of Latin American origin (7:1). In the Navajo Indian population, a prevalence of 1/1,200 has been reported.7 In Mexico, The Registry and Epidemiological Surveillance of External Congenital Malformations has reported a prevalence of 7.37/10,000 newborns (both living and those who died) during the period from 1978-2010, whereas other authors have reported 1:1,500 live newborns.8,9
In tertiary-level care hospitals, this malformation is found among the first reasons for outpatient consults. During the period from 2006-2010 there were 499 cases seen in the Hospital Infantil de México Federico Gómez (HIMFG) and 318 in the Hospital de Pediatría, Centro Medico Nacional Siglo XXI (CMNSXXI). Between 2002 and 2006 there were at least 19 familial cases identified. In the National Rehabilitation Institute there have been 149 cases reported (Departments of Biostatistics and Clinical Records of HIMFG and Dra. María Antonieta Araujo Solís, Hospital de Pediatría, CMNSXXI, Fifth Meeting of Pediatric Research9,10). To contextualize these figures in relation to the high frequency of consultation for microtia-atresia in our population, they can be compared with the frequency reported in the Hungarian Registry of Congenital Abnormalities,11 which identified a total of 980 cases during a 17-year period (1980-1996), with a prevalence of 0.46/1,000 births.
Microtia-atresia most commonly presents unilaterally (79-93%) and on the right side (60%). It occurs predominantly in males and is found to be associated with atresia or stenosis of the external ear canal (55-93%). More than 80% of patients have conductive hypoacusia on the affected side.11-14
A large proportion of patients with bilateral microtia (20-60%) have associated anomalies.6 Congenital defects most commonly seen are vertebral disorders, macrostomia, facial clefts, facial asymmetry, renal abnormalities, heart defects, microophthalmia, holoprosencephaly and polydactyly.1,2,15,16 Many of these abnormalities are also seen in the facio-auricular-vertebral spectrum. Some authors consider microtia to be the minimal expression of this disorder.8,9
Different studies indicate that Mendelian inheritance is more common in syndromic and familial cases, whereas polygenic or multifactorial causes are more probable in sporadic cases. Different risk factors have been described such as the effect of the disorders in the glucose levels in poorly controlled gestational diabetes. There is also evidence that exposure to certain medications such as mycophenolate retinoids and thalidomide cause microtia.17-19 It should be considered that the action of these factors is not unique, but a multifactorial event in which the environment interacts with the genome.
3. Clinical classification
In 1926, Hermann Marx published the first classification system for congenital anomalies of the external ear, which is one of the most used currently.20 Since that time, different classifications have been proposed based on the surgical and embryological appearance of the lesion.21 The majority of clinicians use the Hunter classification system (Fig. 1), which is described below.22
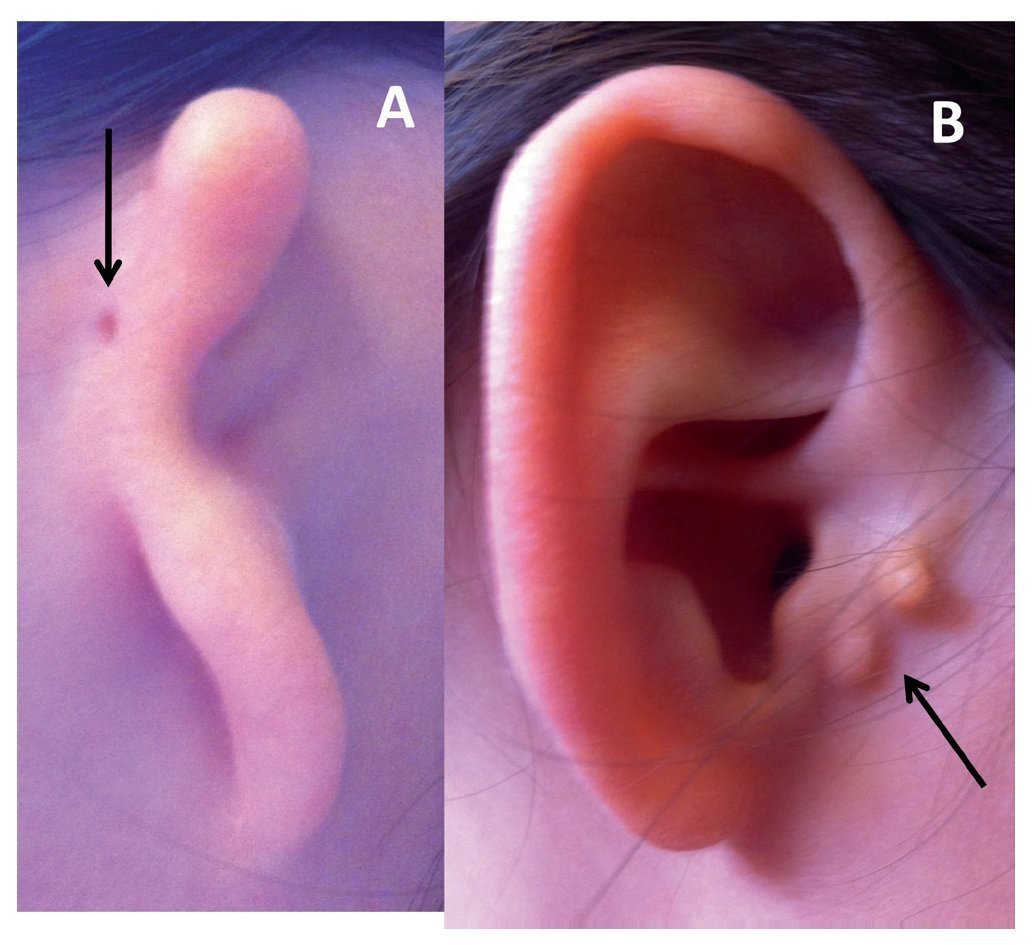
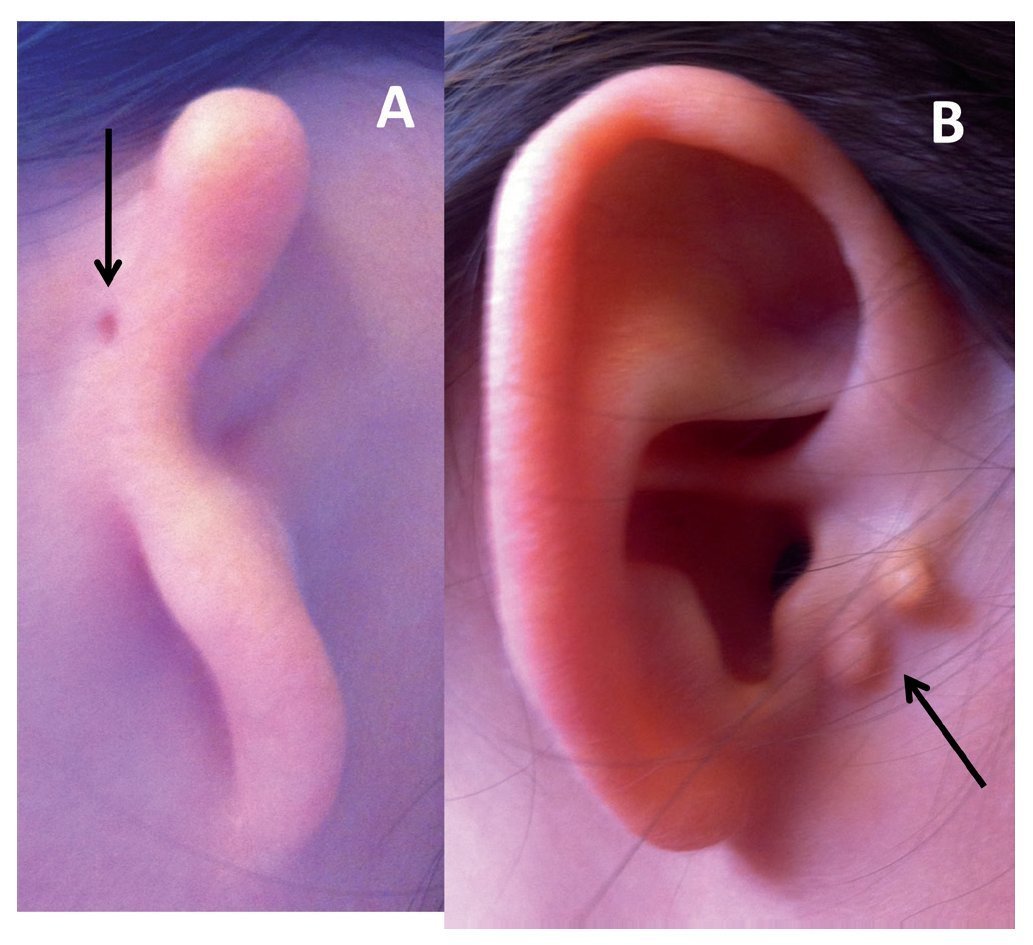
Figure 1 Types of microtia-atresia. A) Type III with auricular skin tag (arrow). B) Type I with preauricular appendices (arrow).
• Type I: Small ear that retains all of its anatomic components, but the length is 2 standard deviations (SD) below the mean
• Type II: Residual tissue of vertical cartilage with presence of some structures of the ear and a length 2 SD below the mean
• Type III: Irregular tissue mass without resemblance to the ear
• Type IV: Absence of the ear
This classification is used by the different specialists who provide care to patients as part of the clinical approach of microtia. Each case is individual and will have its own requirements for care depending on the type of lesion and if it is uni- or bilateral or if it is believed that it can be isolated or is syndromic. According to the judgment of the physician, disorders need to be ruled out at the vertebral, renal and ophthalmological level as well as to carry out hearing tests. Also, in unilateral cases (and if there is a healthy ear), indications must be carefully followed so that the ear remains without damage. It is also important to decrease the risks of frequent infections or exposure to ear toxins caused by some antibiotics that can cause hearing damage that would have been preserved. Once the patient has an optimal health status and growth conditions, a surgical procedure may be considered that would reconstruct the ear. However, we must insist that this is dependent on the case. If it has also been established that there is a hereditary family history or if there was exposure to possible teratogens, etc., genetic counseling should be provided. For these reasons, in these cases the coordinated efforts of pediatricians, geneticists, plastic surgeons, audiologists, ear-nose-throat specialists (ENTs), ophthalmologists and psychologists, among other specialists is required.
4. Embryological basis
The union of the first pouch and pharyngeal cleft with the surrounding tissue of the pharyngeal arches I and II form the structures of the middle and external ear. The pharyngeal arches are comprised of mesenchymal cells of mesodermal origin and cells from the neural crest. The structures of the internal ear arise from the superficial ectoderm. The external auditory canal is formed by the invagination of the pharyngeal cleft, whereas the ectoderm of the cleft forms the epithelium for the duct. The outer ear is formed from six auricular mounds coming from the tissue of the pharyngeal arches I and II. These surround the pharyngeal cleft and contribute to its specific components.23
Initially these structures fuse in the region of the neck and reach the height of the ocular globes due to mandibular growth. Its development begins in the fifth week of gestation and is completed at 12 weeks. Migration of the outer ear to their normal placement occurs at 20 weeks. Different signaling molecules and proteins are involved in the morphogenetic process and in the differentiation of the outer ear.24
5. Association with syndromic entities
Part of the complexity of the study of microtia-atresia is because in only a small number of cases is it possible to identify a purely genetic cause (in monogenic presentations) or purely environmental. In the majority of cases, a multifactorial etiology is established. The one occasionally associated with syndromic entities has important implications with regard to its management, treatment and genetic counseling of the patients.10,25
Among the most common clinical entities in which microtia-atresia may be present as part of the pleiotropic effect of the syndromes are considered the eye-ear-vertebral spectrum, Treacher-Collins syndrome, velocardiofacial syndrome associated with deletion of 22q11.2, among others.
The eye-ear-vertebral spectrum (OMIM 164210)a has a variability of expression that includes the hemifacial microsomy and Goldenhar Syndrome up to the facio-auriclevertebral sequence. This alteration involves the derivatives of the first and second branchial arches and presents craniofacial, cardiac, vertebral and central nervous system (CNS) disorders. It presents unilateral malformation of the external ear and facial malformation of the affected side as well as epibulbar dermoid cysts. Among the genes associated with this syndrome, the GSC gene has been studied and no mutations have been found. However, the BAPX1 gene could cause malformations due to changes in its epigenetic regulation.26 For the most part, these are sporadic cases, but there are familial reports which, in some cases, raise suspicion that the risk may be of an autosomal dominant inheritance pattern. As a consequence, the risk of recurrence would be 50% for children with an affected parent.
Treacher-Collins-1 syndrome (OMIM 154500)a is characterized by downward oblique palpebral fissures, coloboma of the eyelid, micrognathia, microtia, zygomatic hypoplasia and macrostomia. It has an autosomal dominant pattern of inheritance. It is mainly caused by mutation of the TCOF1 gene in 5q32 as well as the genes POLR1D in 13q12.2 and POLR1C in 6p22.3 in 9% of the cases.27-29
Velocardiofacial syndrome (OMIM 192430)a associated with (22q11.2) has >180 clinical characteristics. This deletion has also been associated with the DiGeorge Syndrome and with conotruncal cardiac disorders. Although the typical deletion comprises >30 genes, the gene TBX1, in particular, has been identified as important in the development of the ear.30-32 Although most of the cases presented are de novo, the presence of the deletion should be ruled out in the parents. Given the loss of one of the chromosomal regions of 22q11.2, there is a 50% risk of recurrence for the offspring of an affected parent.
The syndromes mentioned are not the only ones in which microtia-atresia has been reported in a high percentage of the cases. Other examples are the branchio-oto-renal syndrome and the CHARGE syndrome. In a family of Iraqi descent with autosomal recessive segregation, bilateral microtia, hearing alterations and cleft palate have been reported. By means of the analysis of association of the complete genome (Genome Wide Association Study, GWAS), the gene responsible was identified in 7p14.3 where a group of HOXA genes is found. These genes codify for transcription factors and a mutation in the HOXA2 gene has been demonstrated.10
Accordingly, the etiology of microtia-atresia has been related with entities that have an autosomal dominant, autosomal recessive, multifactorial pattern of inheritance as well as alterations in the number of copies of potential genes involved, as suggested by its presence in trisomies 13, 18 and 21, and other unbalanced chromosomal alterations (for example, in the partial deletions of 5p, 18p, 18q and 22q11.2).
Identification of genes related with the presentation of microtia-atresia in syndromic forms is promising because it leads to the consideration that these genes play a role in the preservation of hearing. Changes in these genes or in their interactions can lead to the presentation of the malformation. New technologies are required such as the aforementioned GWAS to identify genes responsible and protein products or regulators that these have during specific periods of embryonic development along with their interactions with the environment and the genome. Therefore, it is important to consider the genetic aspects where the specific genes identified as the responsible individual of some types of microtia-atresia are studied and the genomic aspects where the genome is studied in its entirety to establish the possible causes of microtia-atresia.
6. Genomics of microtia-atresia
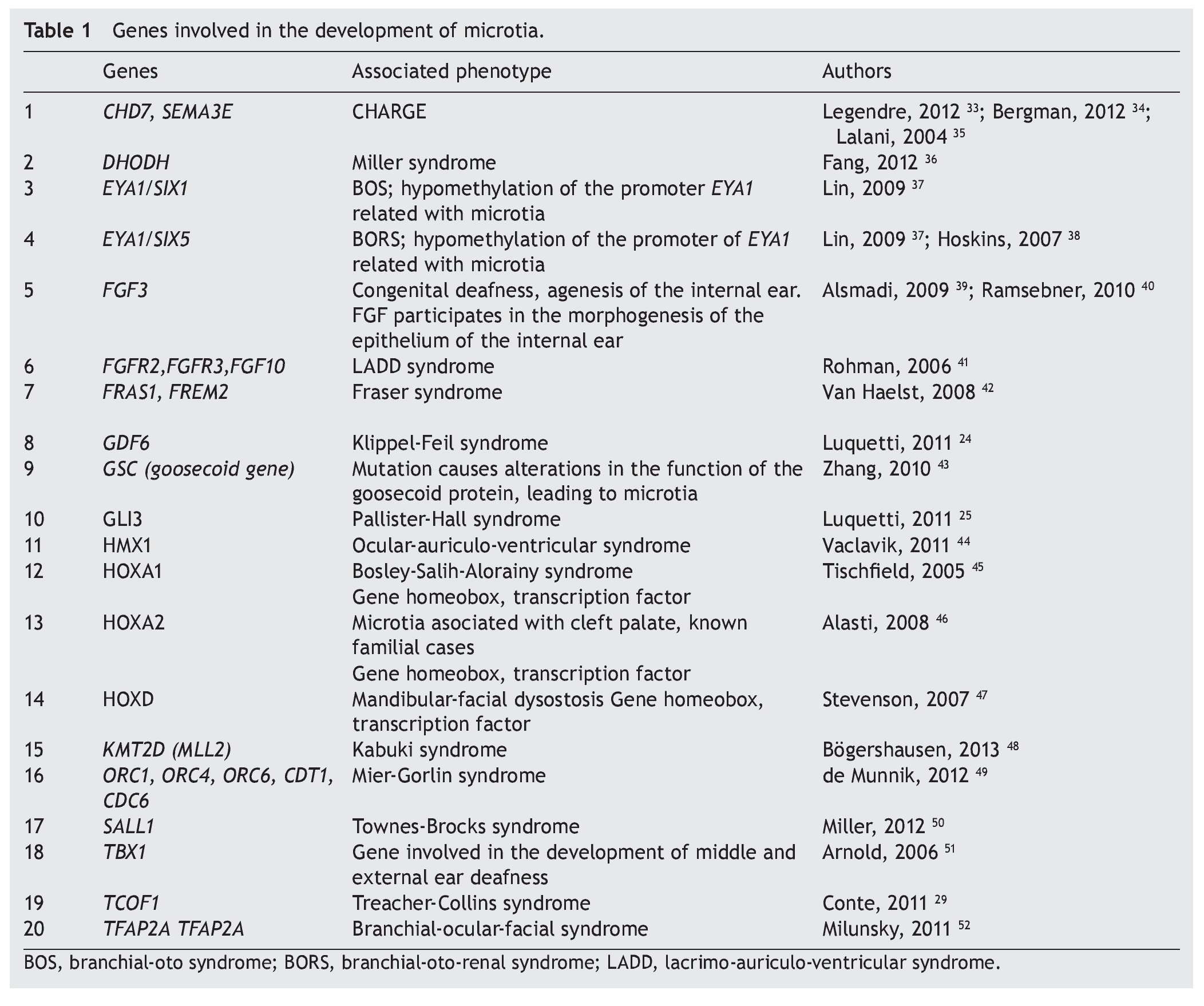
As previously mentioned, microtia/anotia can be found as an isolated entity or associated with syndromes. When it is syndromic, generally it is part of a specific pattern of multiple congenital malformations and the complete entity can be associated with the following factors: 1) exposure to teratogenics such as diabetic embryopathy, various teratogenous agents such isotretinoin, a derivative of retinoic acid, or thalidomide; 2) larger mutations in sole genes, for example, Treacher-Collins syndrome with mutation in the TCOF1 gene (Table 1).34-52 In this context there have been >20 syndromes reported related with the presence of microtia.53 Another factor is 3) the change in the number of copies that can involve trisomies of autosomes, unbalanced structural chromosomal aberrations and pathological CNV (Copy Number Variation).
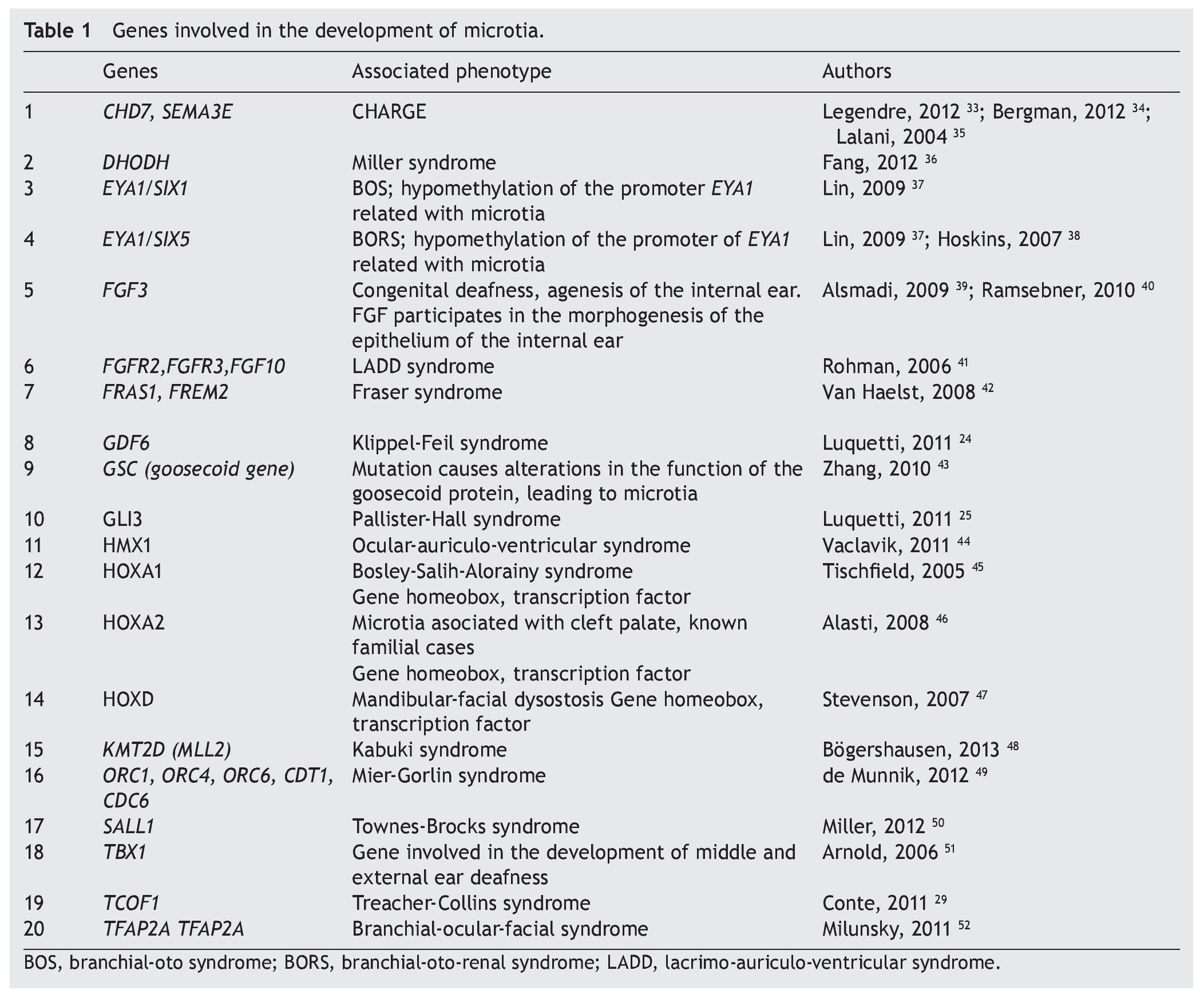
When dealing with mutations of only one gene, there is familial aggregation and different forms of Mendelian inheritance are observed, autosomal recessive, autosomal dominant and X-linked. It must also be taken into consideration that the alterations in any of the genes involved that lead to the generation of microtia are not limited to the change in the sequence of the nucleotides; one gene without mutations could be subject to epigenetic changes that alter its transcription or the alteration could be found in the molecules that regulate the genetic expression at the post-translational level.
In reference to the cases of isolated microtia/anotia, only a low percentage has familial aggregation (4%) and responds to a monogenic pattern of inheritance. Accordingly, it is proposed that isolated microtia/anotia responds primarily to a pattern of multifactorial inheritance involving multiple genes of lesser influence in addition to environmental factors that alter the threshold of normality.54 A proposed environmental factor for the development of microtia has been altitude >2,000 m above seal levels,53 mainly based on the high prevalence of microtia in Latin American populations53-57 especially in cities such as Quito, La Paz and Bogota. However, prevalence has not been reported in any other cities with high altitudes such as Tíbet, for which it is feasible that ancestral Amerindian inheritance could also have an influence in the high prevalence of this malformation.
6.1. Development of the external ear is orchestrated by multiple genes
A large number of syndromes demonstrate microtia,23,53 which calls attention that many genes intervene in the development of the external ear. To date, all genes that play a role in these syndromes have not been identified; however, there is evidence that some are involved in the morphogenetic processes of development of the ear (Table 1). An important group of genes with a primordial role in the development of the ear are the homeotic genes such as SIX, HOXA1, HOXA2 and HOXD, which are factors of transcription and therefore regulate the activity of other genes throughout the genome.10,25,58 One of the known functions is to establish spatial or positional identity of different embryonic structures, especially on the formation of the anterior-posterior pattern of formation of the embryo. These genes are very important in ontological human development because they function by regulating a not yet established number of genes that are their transcriptional targets. The HOXA2 gene, especially directly related with microtia,46 is a transcription factor that acts as a gene selector that will express in the morphogenesis of the neural crest and in the second branchial arch, structures that give rise to the formation of the ear.10,24,58,59 In fact, Brown et al., using exome sequencing techniques, described a family in which bilateral microtia and hearing loss were segregated with the nonsense mutation c.703C>T, p.Q235* in HOXA2.60
The number of genes implicated in the development of the external ear and the fact that their dysfunction could cause microtia increases because the HOX genes, in turn, could be regulated by another type of genomic component such as microRNA (miRNA). The miRNA are small molecules of non-codifying simple chain RNA of ~22 nucleotides that regulate the protein synthesis by degradation of messenger RNA (mRNA) or by repression of translation.b miRNA are essential elements during the embryonic development where they are expressed in tissue-specific manner regulating the differentiation, the establishment of patterns and the morphogenesis. It is known that mutations in MIR96 could be related with nonsyndromic deafness.61 In addition, the HOXA2 gene has been proposed as a target for ~30 miRNA (mirDB); and the HOXA1 gene is the proven target of miRNA hsa-miR-10a (TarBase).c
Recently, Li et al.62 found a differential expression of miRNA in cartilage and soft tissue of the external ear in 58 pediatric patients with microtia in comparison with the tissue of the external ear in control individuals. They found over-expression in six miRNA (miR-486-5p, miR-451, miR-140-3p, miR-16, miR-185 and miR-126) and a low expression of five miRNA (miR-708, miR-1308, miR-200c, miR-203 and miR-205) in patients with microtia. When the target genes of these miRNA were studied, the target gene of miR-200cTRPS1 (zinc finger transcription factor Trps1) was underexpressed. As miR-200c has a low expression, it is interpreted that the direct target of this miRNA is a repressor of TRPSI which, in turn, is a transcriptional regulator gene with specific union to the GATA sequence. When TRPS1 acts, it represses the transcription of genes that have been implicated in multiple functions and in the proliferation of chondrocytes, important cells in the development of the external ear, as abnormal development of the cartilage is a central characteristic in microtia. Other target genes of miR-200c are OSR1 (Odd-skipped related 1) gene related with the development of intermediate mesoderm and branchial arches during embryogenesis and GLI3 associated with the Pallister-Hall syndrome with microtia.25,62 Although it has been studied in a cohort of live born pediatric patients, this proves that the participation of miRNA in the formation of the outer ear is important and that its alteration could cause microtia. An important observation is that miRNA are also subject to the CNV, adding complexity to the underlying genome related to the appearance of microtia.b
6.2. Microtia is associated with unbalanced genomes
Genomic imbalances come in different sizes. They can be as large as to affect entire chromosomes, as in aneuploidies, or so small as to not even be detected microscopically as with some submicroscopic copy number variants (CNV) and can be defined as a segment of DNA from ≥1 kb that is present in a variable number of copies compared with a reference genome. A CNV could be simple in its structure such as tandem duplication or could imply complex gains or losses of homologous sequences found in multiple sites in the genome. CNV could influence gene expression by interrupting genes or altering the gene dose. Some examples are syndromes of microdeletion or microduplication but also can be associated with complex characteristics or diseases.63
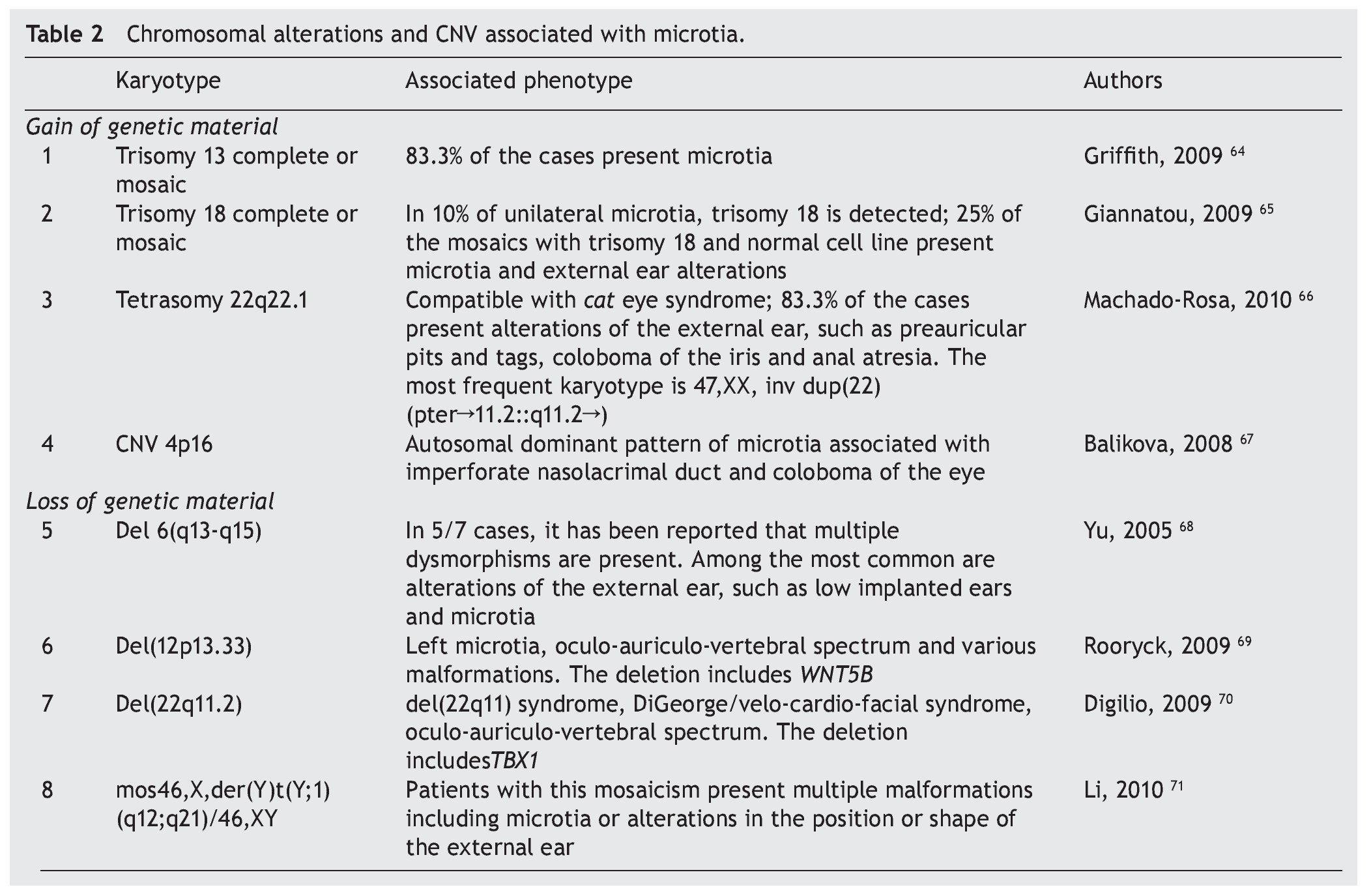
In patients with microtia, in addition to the main genes and their regulators, syndromic cases are commonly associated with changes in the copy number such as duplications or deletions, which is found in virtually every human chromosome (Table 2)64-71 suggesting that the basic defect is in a pathway of organogenesis.
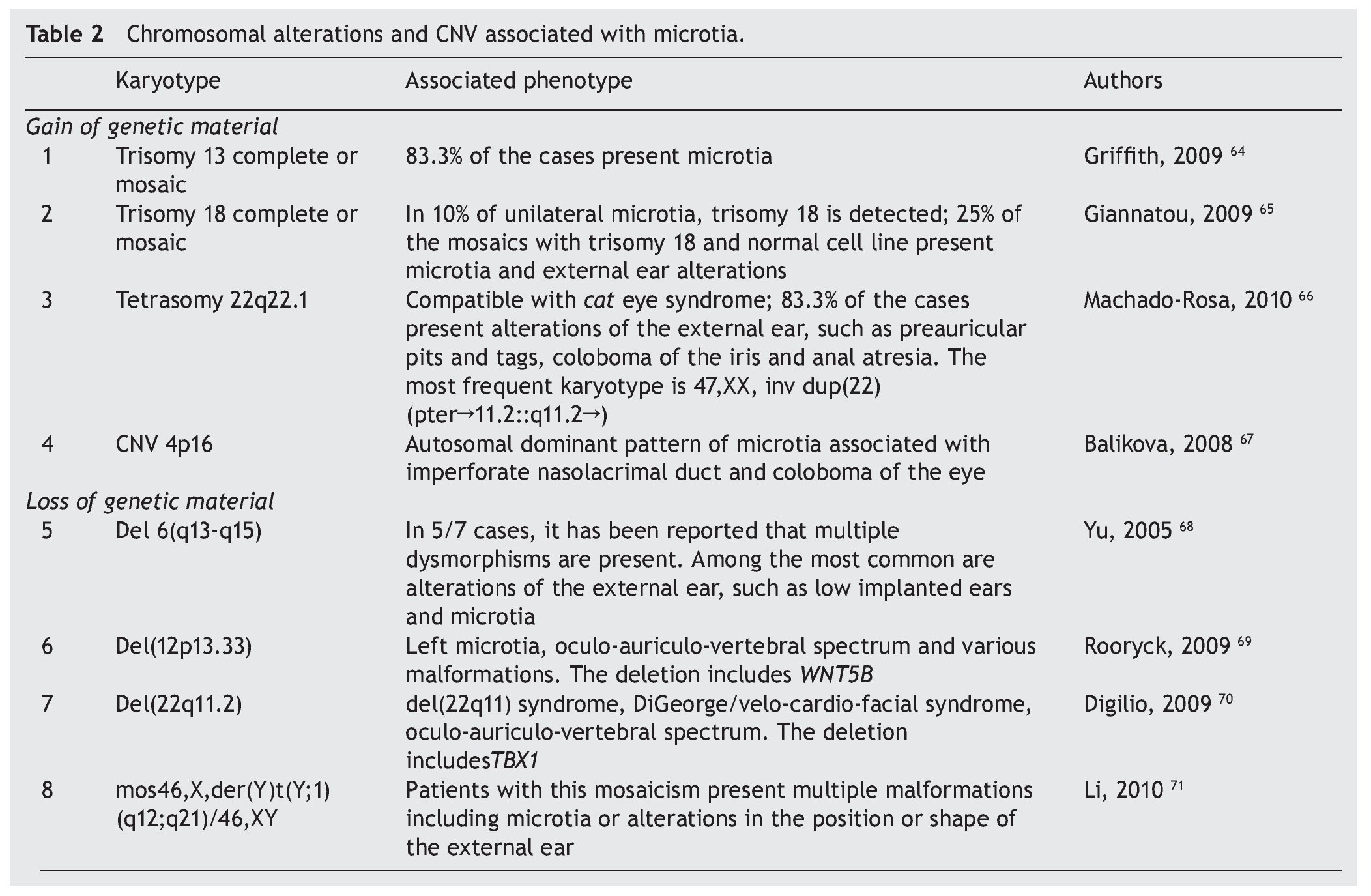
Microtia has been part of the clinical picture in the most common aneuploidies such as trisomy 13 and 18.25 When a series of patients with microtia/anotia have been cytogenetically studied, a variable percentage of alterations have been found. For example, in 172 cases of microtia/anotia, Mastroiacovo et al. found ten of syndromic type, of which two cases had trisomy 13 and four cases had trisomy 18.16,64
Similarly, Forrester and Merz found four cases with trisomy 18 in 41 patients with microtia.4 In cases of mosaicism of trisomies 13 or 18 with normal cells, microtia has also been found in a high percentage of patients.65,66
In partial imbalances (incomplete chromosomes), microtia/anotia has been reported repeatedly in various unbalanced alterations. Examples of this are the cat eye syndrome,66 which is caused by a partial tetrasomy of chromosome 22 with the recurrent karyotype 47,XX,+inv dup(22) (pter#fhaciaderechaq11.2::q11.2#fhaciaderechapter) and in diverse partial deletions of the chromosomes 6, 12 and 22. This suggests that the haploinsufficiency of genes located in the involved regions is related with the generation of microtia. However, the presence of a large number of copies in some genetic regions also can give rise to microtia, as observed in trisomy 13 and 18, but also by the presence of CNV consisting of five copies of a 750 kb amplicon located on the short arm of chromosome 4. This CNV segregates with microtia and other malformations in an autosomal dominant inheritance mode.
In summary, information about the association of microtia with mutations or alterations in the amount of major and minor genes and their regulators such as miRNA reveals that there are a large number of loci required in the normal formation of the outer ear. For this, it can be considered that its development is the result of a concerted genomic activity in quantity, time and space of various genes and environmental factors that must act harmoniously for normal organ development. Some failure in genomic or environmental factors or their interactions could cause microtia.
As shown, microtia-atresia is a malformation of great significance for a variety of health services in Mexico because of the different areas and specialists involved that includes but is not limited to pediatricians, plastic surgeons, audiologists and speech therapists, otolaryngologists and medical geneticists. It is important that professionals working with these patients be aware of the clinical, molecular and hereditary bases of the disease. Although there is a growing interest in relation to this disease, there are still important issues to be elucidated in relation to the genetic, genomic, and proteomic aspects in this malformation of high prevalence in our country.
Conflict of interest
The authors declare no conflict of interest of any nature.
a Online Mendelian Inheritance in Man. Available at: http://www.ncbi.nlm.nih.gov/omim
b miRBase: the microRNA database. Available at: http://mirbase.org/
c DIANA Lab. DNA Intelligent Analysis. Available at: http://diana.cslab.ece.ntua.gr/tarbase/
Received 31 July 2014;
accepted 5 November 2014
☆ Please cite this article as: Aguinaga-Ríos M, Sara Frías, Arenas-Aranda DJ, Morán-Barroso VF. Microtia-atresia: Aspectos clínicos, genéticos y genómicos. Bol Med Hosp Infant Mex. 2014;71:377-386
* Corresponding author.
E-mail: vfmoran@himfg.edu.mx (V.F. Morán-Barroso).