



Acute leukemias have a huge morphological, cytogenetic and molecular heterogeneity and genetic polymorphisms associated with susceptibility. Every leukemia presents causal factors associated with the development of the disease. Particularly, when three factors are present, they result in the development of acute leukemia. These phenomena are susceptibility, environmental exposure and a period that, for this model, has been called the period of vulnerability. This framework shows how the concepts of molecular epidemiology have established a reference from which it is more feasible to identify the environmental factors associated with the development of leukemia in children. Subsequently, the arguments show that only susceptible children are likely to develop leukemia once exposed to an environmental factor. For additional exposure, if the child is not susceptible to leukemia, the disease does not develop. In addition, this exposure should occur during a time window when hematopoietic cells and their environment are more vulnerable to such interaction, causing the development of leukemia. This model seeks to predict the time when the leukemia develops and attempts to give a context in which the causality of childhood leukemia should be studied. This information can influence and reduce the risk of a child developing leukemia.
La leucemia aguda representa una enfermedad con una enorme heterogeneidad morfológica, citogenética, molecular y polimorfismos genéticos asociados con la susceptibilidad. Presenta factores causales asociados con el desarrollo de la misma. Particularmente, cuando tres fenómenos se conjuntan, traen como resultado el desarrollo de la leucemia aguda. Estos fenómenos son la susceptibilidad, la exposición ambiental y un periodo que, para este modelo, ha sido denominado el periodo de vulnerabilidad. El presente marco teórico muestra cómo los conceptos de la epidemiología molecular han permitido establecer una referencia a partir de la cual es más factible identificar los factores ambientales relacionados con el desarrollo de la leucemia en niños. Posteriormente se muestran los argumentos para predecir que solo los niños susceptibles probablemente desarrollarán leucemia una vez que se exponen a un factor ambiental. Por lo que, por más exposición, si el niño no es susceptible a la leucemia, no la desarrollará. Además, esta exposición debe ocurrir durante una ventana de tiempo durante el cual las células hematopoyéticas y su entorno son más vulnerables, para que dicha interacción provoque el desarrollo de leucemia. Este modelo pretende predecir el momento en el cual se desarrollará la leucemia y trata de dar un contexto en el que la causalidad de la leucemia en niños deberá ser estudiada. A través de esto se podrá influir y disminuir el riesgo de que un niño desarrolle leucemia.
This model aims to lay the groundwork to identify factors associated with the development of acute leukemia (AL) in children. First, the concepts of molecular epidemiology are explained, which are those that give rise to this theoretical model. Subsequently, the components of the model are described and, finally, a description of the same is done.
2Molecular epidemiologyFor Shulte, molecular epidemiology emerges as an evolutionary state of the epidemiology in which it will not only be important to identify the risk factors of the diseases. The mechanisms that lead to their development can be identified through molecular epidemiology. From this point of view, it will result in the emergence of new theories and, with it, the maneuvers for disease prevention in the population can more accurately be directed.1 The Shulte concept has proved some reactions: some in favor,2–15 others against16-18 and others, although they do not discard molecular epidemiology as a new discipline question it and expect its prompt strengthening.19-23Table 1 shows the most common definitions of molecular epidemiology.14
Evolution of the molecular epidemiology definition.
| Author (s) | Year | Definition |
|---|---|---|
| Higginson J | 1977 | “The application of sophisticated techniques to the epidemiologic study of biologic material” |
| Schulte PA | 1993 | “Molecular epidemiology is the use of biologic markers or biologic measurements in epidemiologic research” |
| Tompkins LS | 1994 | “The application of molecular biology to the study of infectious disease epidemiology” |
| McMichael AJ | 1994 | “Using molecular biomarkers in epidemiology” |
| Groopman JD, Kensler TW, Links JM. | 1995 | “Molecular epidemiologic research involves the identification of relations between previous exposure to some putative causative agent and subsequent biological effects in a cluster of individuals in populations” |
| Hall A | 1996 | “The analysis of nucleic acids and proteins in the study of health and disease determinants in human populations” |
| Shpilberg O, Dorman JS, Ferrell RE, et al. | 1997 | “Molecular epidemiology uses molecular techniques to define disease and its pre-clinical states, to quantify exposure and its early biological effect, and to identify the presence of susceptibility genes” |
| Levin BR, Lipsitch M, Bonhoeffer S. | 1999 | “The practical goals of molecular epidemiology are to identify the microparasites responsible for infectious diseases and determine their physical sources, their biological relationships, and their route of transmission and those of the genes responsible for their virulence, vaccine-relevant antigens and drug resistance” |
What allows us to speak of a new discipline are not only the techniques or tools applied in the epidemiology, but the concepts that this brings. These concepts are the internal dose, the effective biological dose, early biological effects and the altered function and structure, concepts that are made operational though the different biomarkers.1 In this sense, the biomarkers can be divided into biomarkers of exposure, effect and susceptibility.1 Exposure biomarkers are those of internal dose, biologically effective dose and target tissue dose. Susceptibility biomarkers include polymorphisms in genes involved in the metabolism of carcinogens, in DNA repair and in the control of the cellular cycle. Biomarkers of effect evaluate the early genetic alterations and the modulation of the nutritional and immunological state that lead to tumorigenesis.4
Molecular epidemiology has been proposed as an evolving state of the classic epidemiology, which for some only represents the incorporation of new techniques, the molecular techniques, in epidemiological designs,16,20 whereas for others it represents the best way to explain the mechanisms related with the health-disease processes in human populations.1,4 This provides the answer to the problem of epidemiology that, on establishing an association between an environmental factor and disease, leaves a “black box”; i.e., that cannot clarify the mechanisms by which this factor causes the disease.1 In such a case, the molecular epidemiology would contribute to the solution for this black box.2,24
However, molecular epidemiology does not only arise as an incorporation of new molecular techniques, as Shulte himself proposes. It arises from the identification of individuals who, despite being exposed to the same risk factors, do not present the same response, whether it is because they do not receive the same dosage (internal dose, effective biological dose) or because they respond metabolically in a different manner to the substance (susceptibility biomarkers). It also arises from the concern of identifying in a timely manner the damage caused by a toxic agent (effect biomarkers). This leads to the development of molecular epidemiology.4,7,14
It is then clear that in any epidemiological investigation it is possible to apply molecular epidemiology, although not necessarily. The principal level of analysis of epidemiology is population level, and its principal objective, as part of public health, is prevention.18 Epidemiology achieved the biggest advance with regard to cancer and cardiovascular disease prevention by establishing a relationship between these diseases with tobacco consumption without the need for molecular epidemiology.25-28 It is not the fact that most diseases in humans are multicausal, which determines the emergence of molecular epidemiology,1 or fallen into an environmental reductionism in search of an only risk factor for identifying causality of a disease.2 Identification of smoking as a sole environmental factor associated with lung cancer or with coronary disease offered an important answer to the causality and prevention of these diseases.27-29 Almost 80% of the different types of lung cancer and 50% of ischemic heart diseases are caused by tobacco.25,30,31
Molecular epidemiology emerges as an alternative when it is not possible to identify environmental factors of disease in the population and when it has been demonstrated that a substance is potentially harmful at the individual or basic levels.32 This could be the result of errors in measurement of the exposure or it may be that some individuals have different responses to a particular substance.4,32 In those cases there is no other alternative than molecular epidemiology.1,3 To use molecular techniques in associations shown by epidemiology results in the accumulation of further evidences of the epidemiological association.1,28 However, it is also likely that there is a fetishism of the indiscriminate use of the technology that may lead to the thinking that with the advanced technology one may come to a greater understanding of the biological phenomenon, which could be an error.33 You can continue to demonstrate that tobacco causes lung cancer, and pass all levels of measurement, move from the population level to the individual level, and from there to the molecular;34 however, this is not what allows explaining the health-disease process.34 On the contrary, many times this only generates hierarchies between disciplines and a false idea of the greater strength of the finding. It is believed that it is explained further when all that is achieved is a description at a lower level of measurement in a more reductionist vision of the phenomenon.35 Finding traces of derivatives of tobacco in malignant lung tissue is not evidence of the association between tobacco and lung cancer, but a retrograde vision of single cause theory of Koch’s postulates, which are unable to explain the health/disease process in human populations.36
Epidemiology has a predominant value in public health that does not depend on the techniques used to substantiate its findings. Neither the causality of diseases nor their prevention depends on the techniques used.18 Snow, Doll and Hill did not use great techniques to prevent the diseases they studied.27,37 Causality depends more on the value judgments than on the techniques used.38,39
Molecular epidemiology is a branch of epidemiology and, for some in particular, a branch of clinical epidemiology.1 It is not an evolving state where what evolves is better or stronger than what it leaves behind.1,20 It is a branch that will develop in parallel to the trunk; public health develops and incorporates new concepts and techniques to its duties.18,21
The advantage of molecular epidemiology is then related to the dosages that are really absorbed (internal dose) and to the dosages capable of producing an effect on the organism (effective biological dose), events that are not measured and many times not even considered by classic epidemiology. On the other hand is the evaluation of the interaction between environmental exposure and genetic polymorphism, which intervenes in the metabolism of toxins. Finally, molecular epidemiology is able to identify the initial damage caused to the body by a substance or agent; therefore, it is feasible to reach the early identification of the disease.1,4,32 Without molecular epidemiology it would not be possible to achieve all of this.32
One of the most promising fields within molecular epidemiology is to identify the interaction between environmental exposures and susceptibility of the individual as a way of identifying those persons truly at risk7 and, on the other hand, to identify factors that had not been detected as carcinogenic in population studies.4,11 The finding of associations between an exposure and disease could be more noticeable in a particular genetic polymorphism28 or in individuals susceptible to the disease.7 The study of the population at greater risk of suffering from a disease could be a good model to predict what would happen to the general population.1,4 At the least, factors that could increase the risk of suffering from a disease will be able to be identified.4
Finally, the limitations of molecular epidemiology should be made clear.21 It has been long known that the dose does not necessarily reflect the degree of involvement in an individual, especially when we are dealing with infectious diseases.36 It is also important to point out that the effective biological dose is not necessarily an indicator that the disease will progress, given that extensive damage to the cells is reversible.40 Not even the declared states of diseases are a guarantee that there may not be an involution of the disease.41 Doubt then arises as to whether it would always be advantageous to carry out an early diagnosis given that in some cases there is the possibility of an involution of the phenomenon.40,41 Another major problem of early diagnosis is that there is not always the possibility of providing appropriate treatment to the patient.42 It should also be mentioned that the search for associations between exposure to carcinogens and genetic polymorphisms of the genes that intervene in the metabolism of the carcinogens have offered contradictory information to the point that it would appear that this is not the path that explains why some individuals develop the disease and others do not.43,44 Finally, identifying the population at risk is not always advantageous for the prevention of disease42 because factors that cause the population at risk to develop the disease are not yet known. On the contrary, the person can develop a degree of anxiety by knowing that he/she is a carrier of a gene that can increase risk of suffering from a disease, yet cannot be counseled as to what exposures should be avoided.18 Another aspect that should not be forgotten is that using biological material for diagnosis, such as the search for related genes, causes a decrease in the participation of the population or the power to capture the eligible population, which itself can produce the presence of selection bias.21,23 One cannot then leave aside that molecular epidemiology faces the same problems as classic epidemiology and that the use of molecular techniques does not guarantee that the best markers are being used for the measurement of the exposure.21 It should be kept in mind that not all biomarkers have been validated.21,22 Use of the technology does not prevent the presence of selection bias21,23 nor does it guarantee that the bias of confusion can be eliminated.21
Despite its limitations, molecular epidemiology is an accepted discipline that permits the identification of why certain environmental factors in some populations produce disease and others do not.24 Molecular epidemiology is also a good example of interdisciplinary research.1,4 There are two types of interdisciplinary research: that where two or more disciplines unite their techniques to answer the same research question and another when two or more disciplines merge their concepts to answer a research question. The latter is also known as trans-discipline.45 Further examples of molecular epidemiology are where the only result that has occurred is an exchange of techniques between molecular biology and epidemiology.16,21,46 What can be more difficult is to reach a trans-discipline where the disease can be explained from the molecular biology and epidemiology point of view. Whereas for one discipline the most important thing is what occurs in the population, for the other the most important is what occurs in the molecules. Each discipline has different ways of interpreting reality given that there are different concepts.21 However, despite there being only one intersection currently at the technical level, it is to be hoped and expected that a mixture of concepts exists that allow for the health/disease process to be explained from the molecular level up to the population level.21,22,46–48 An example of this is the model that attempts to explain where there is an intersection, from the molecular environment, in the development of leukemia in children.49
3Acute leukemia as result of the interaction of different phenomena3.1ExposureWhen mentioning exposure as a phenomenon, it should be pointed out that there are evidences that the exposures cause AL in children. However, one cannot speak of a single exposure as there are different types. The most consistent in the development of AL is in utero exposure to radiation.50 Occupational exposures follow in consistency,51–54 especially those that have to do with the use of petroleum derivatives.54 However, there are other types of exposures, such as biological. Such is the case of the infectious agents,55,56 chemical exposures such as food contaminated with pesticides57,58 or the use of medications such as chloramphenicol.59 One aspect that should be stressed is that very few cases of AL can be explained by currently known exposure factors or by one alone.49
Although some risk factors for developing AL may be high, in the case of paternal occupation the odds ratio can vary from 2-5.51 In fact, few cases can be found associated with each factor in particular.54 What could the child have been exposed to in order to develop AL? It could have been x-ray exposure during pregnancy, an infectious agent during the first 2 to 3 years of life, a toxic agent in the food or environment, electromagnetic fields, different agents in the work environment of the father or mother, etc.49. Specificity of the exposure may not be so important because factors involved with the development of AL may be associated with other diseases. For example, x-rays are associated with thyroid and other types of cancer;60 tobacco consumption by the parents and exposure to hydrocarbons are associated with lymphomas;61,62 and electromagnetic fields and residing close to areas with high-density traffic with tumors of the central nervous system.63 Perhaps some exposure may be specific for AL, such as the case of the human T-lymphotropic virus type 1 (HTLV1) virus in T-cells AL;56,64 however, this is not the rule. That specificity may perhaps have more to do with the child’s susceptibility and with the time in which the exposure takes place. This could also explain why in many children who have been exposed to the same factor, only some develop the disease.
3.2SusceptibilityThe most common susceptibility factor linked to the development of AL is Down’s syndrome. Other syndromes are associated with a high risk of developing AL such as Li-Fraumeni syndrome,65,66 Bloom syndrome, Fanconi anemia, Blackfan-Diamond syndrome and Schwachman-Diamond syndrome, among others.65 However, these conditions represent a small percentage of the total number of cases with AL.65 In utero exposure to x-rays and Down’s syndrome represent only 10% of the AL cases.50 Current molecular techniques have allowed for identification of an enormous quantity of molecular variations that predispose the development of AL, although it has not been possible to arrive at a specific treatment in all cases.67
Another factor that also increases susceptibility is the human leukocyte antigen complex (HLA; also known as major histocompatibility complex or MHC in mice). It is known that the association between an HLA allele and an oncogenic factor could trigger the development of leukemia.68,69 An exaggerated response may exist to a common infection and this response may be regulated by the HLA system.69 In addition, depending on a specific HLA allele, some viruses may not be recognized as foreign by the organism and circumvent immunological defense barriers against the cancer.69,70 This latter factor itself could be the basis for development of AL. However, from the point of view of Greaves,71 when the child does not have an early infection (before 1 year of life) the response provoked in the immunological system when an infection occurs later on would be much more aggressive and trigger a second mutation. This may have as a consequence development of ALL (acute lymphoblastic leukemia). Greaves and Chan point that the mutation is not necessarily germinal, but that mutations could be spontaneous. During the first year of life, it is estimated that there are 1.8 × 1013 pre-B lymphocytes produced, which increases the probability of producing a pre-B cell with a spontaneous mutation.72 If, in addition, between 2 and 4 years of age an infection occurs, the biological marker is created in which ALL could develop. This suggests the participation of HLA. If a late infection occurs (particularly viral infections) in the child who experienced a spontaneous mutation and, in addition, the HLA does not allow for the attack against this infectious agent to be regulated.56 This would generate an exaggerated response against the infection and provoke the second mutation required for the development of ALL.69,70 Therefore, evaluating the HLA in the child with Down’s syndrome would have great importance. The non-susceptible population (without Down’s syndrome as a population control) has the same probability of having the risk allele (or protection) of HLA as a child who does develop ALL or another type of AL because these controls may not have experienced the first mutation. A mutation already exists in the child with Down’s syndrome. Although trisomy 21 or the alterations that may arise in other genes would not be the factors that directly predispose to AL, a low capacity of DNA repair exists73 and there would be a higher risk of presenting a spontaneous mutation in pre-B cells. Similarly, if there is a spontaneous mutation plus the presence of the HLA risk allele (or the absence of one of protection) when an exaggerated response to an infection occurs together with an exposure to another factor that has the potential for causing leukemia, the child could develop the disease.
Chan and Greaves71 and Chan72 proposed that the susceptibility for ALL is more acquired than germinal, as offered by the Knudson theory or the Gardner hypothesis for explaining the cases of ALL that appeared in Sellafield. In this report, Gardner et al. note that there is a mutation that the parents transmit to their progeny,74 even though this proposal has been widely criticized.75-77 Within that acquired susceptibility, one could also include dietary causes (deficiency) and even stressors.59 These two last factors warrant further investigation. Considering the limitation of establishing an adequate causal relationship, they could cause a certain susceptibility both for carcinogenic substances (as is the case that some nutritional deficiencies are associated with a greater effect of radiation) as a poor immune response against cancer cells.78 These factors, however, act at a specific time of the child’s life and may be considered that the predisposition is only momentary. This is of great importance because a child with a high susceptibility for AL, as is the child with Down’s syndrome, on being exposed to a potentially leukemogenic factor, could develop AL if the exposure occurs at a time in which the susceptibility to that factor increases. Consequently, for two children with Down’s syndrome who are exposed to the same risk factor, what can determine that one develops AL and not the other is the time of greater susceptibility.70,79
3.3Period of vulnerabilityFinally, one must consider that this “time of increased susceptibility” not only can determine a normal or abnormal response to an infection where the B cells might be more susceptible to mutate or a deficiency of some particular nutrient or some stress factor. This time could also be a biologically determined time. The peak age of ALL (especially common ALL or CD10 positive) is between 2 and 4 years, whereas AML (acute myeloid leukemia) does not have a peak age in those <15 years of age.80 Greaves notes that if a spontaneous mutation occurred during the first year of life, it is during this age when a child is more predisposed to develop a second mutation in response to an infection.72,81 If this hypothesis is set aside and it is considered that during the first 3 years of life there is a great production of cells from the immune apparatus, especially pre-B cells82 when there is a high level of proliferation found, this alone would increase the effect of any carcinogen, from an infectious one to a chemical exposure.83-85 Accepting that during this period in which the child becomes most vulnerable to carcinogens is a biologically determined time where the tissue or organ affected is found to be in greater proliferation, this would explain the peak ages in which the different neoplasms occur.49,80,86 However, this biological time of greater vulnerability would only explain cancer cases that appear during peak ages, whereas the momentary susceptibility could explain the cases that appear outside the peak age. For example, if an 8-year-old susceptible (Down’s syndrome) child suffers an infection by an agent not well recognized by the HLA or causes an abnormal response or simply provokes a greater proliferation of B cells, the infection if caused by an oncogenic virus could be sufficient to cause a neoplasm or render the child vulnerable for the effect of another oncogenic factor (X-rays, electromagnetics, etc.), finally leading to the development of AL (Figs. 1 and 2). Summing up, it can be said that the factors related with the causality of AL are three: exposure, susceptibility and time. These three phenomena maintain a narrow equilibrium. Therefore, in a society where the susceptible population increases along with the exposure factors, the frequency of AL will increase. Only if it is better understood how a child develops leukemia, the disease may be prevented.87

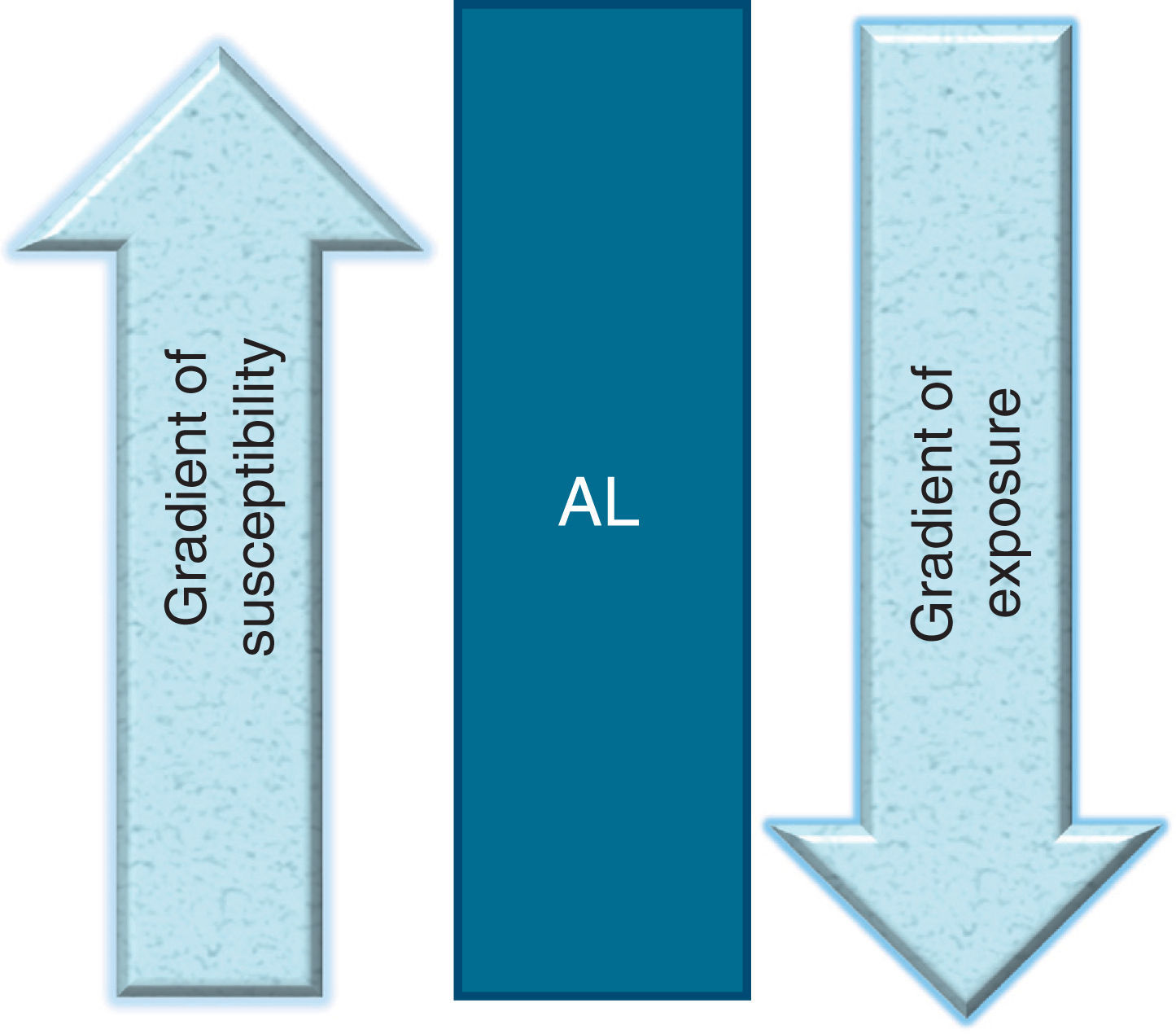
What is required for an acute leukemia to be developed in children? An inverse relationship between the degree of exposure to environmental factors and the degree of susceptibility: the more exposure a child has, the less susceptibility to develop leukemia. The more susceptible the child, the less exposure required for developing leukemia. AL, acute leukemia.
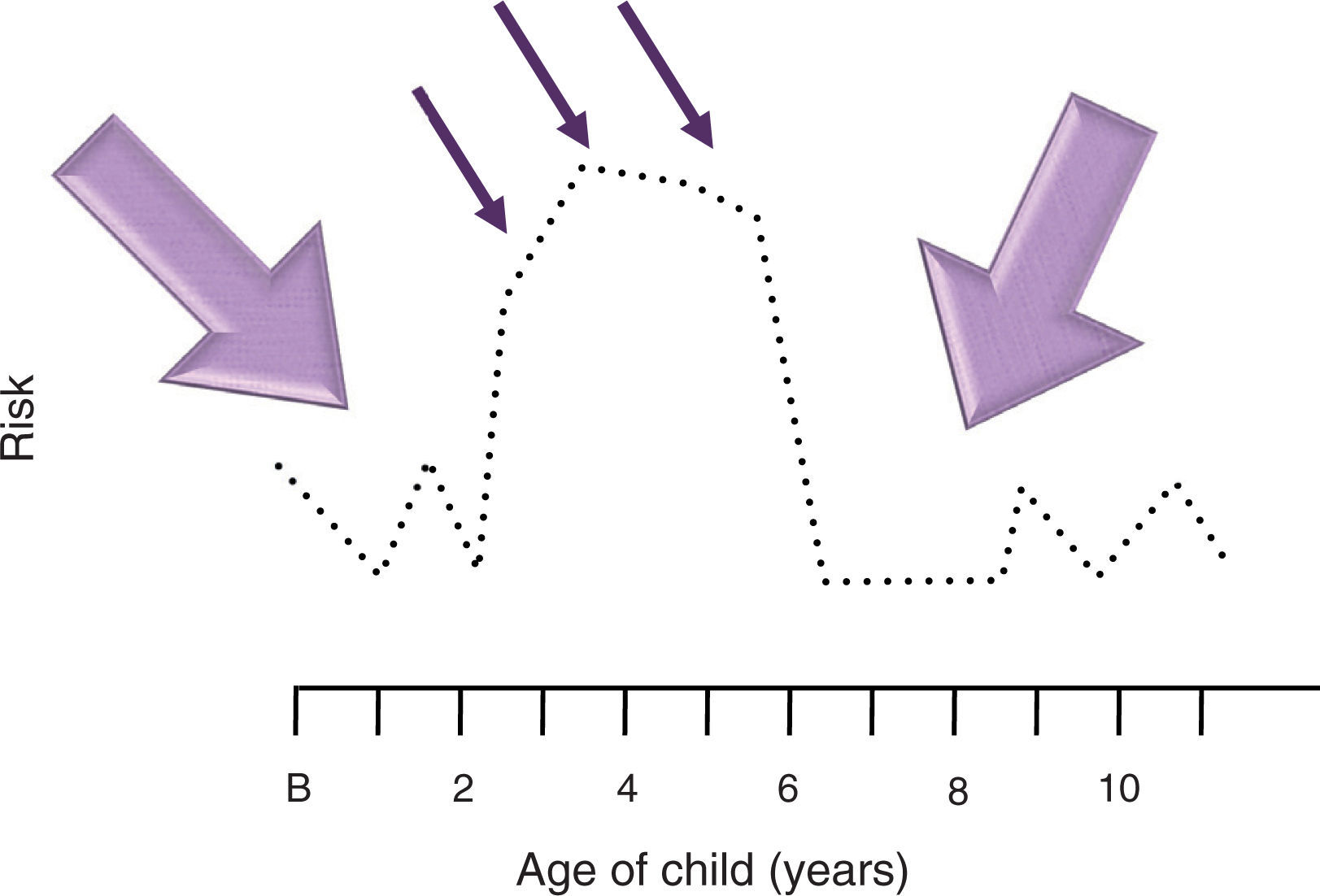
 this period of natural vulnerability occurs between 2 and 6 years of age, a time in which the peak age of the leukemias appears. When the interaction between the exposure and the susceptibility occurs during this period of life, the risk of a child developing ALL increases. This scheme seeks to illustrate that if susceptibility is increased for a few years in children who will develop ALL (illustrated with dotted line), for example, between 2 and 6 years of age, they would require little exposure (thin arrow) to develop leukemia. On the other hand, during the period when susceptibility is low, more exposure would be necessary (thick arrow). B, birth.")
Model of interaction between exposure, susceptibility and vulnerability. A child requires being susceptible to leukemia so that an environmental factor could cause the development of acute leukemia. The same occurs in an inverse fashion. However, this interaction potentiates when there is a significant degree of proliferation in hematopoietic cells that allows for acute leukemia to be produced. In this model, it is called the period of vulnerability. In acute lymphoblastic leukemia (ALL) this period of natural vulnerability occurs between 2 and 6 years of age, a time in which the peak age of the leukemias appears. When the interaction between the exposure and the susceptibility occurs during this period of life, the risk of a child developing ALL increases. This scheme seeks to illustrate that if susceptibility is increased for a few years in children who will develop ALL (illustrated with dotted line), for example, between 2 and 6 years of age, they would require little exposure (thin arrow) to develop leukemia. On the other hand, during the period when susceptibility is low, more exposure would be necessary (thick arrow). B, birth.
To identify the origin of different biological processes in disease is difficult due to the complex interaction among the variables involved in disease onset. This holds true for ALL in children. In recent years, it has been possible to identify different aspects concerning the molecular biology of disease, with several genetic susceptibility factors having been identified for childhood leukemia. However, less successful has been the search for causal environmental factors.
There does not seem to be a clear answer as to why children become sick with ALL. At present, it is accepted that ALL is the result of the interaction between susceptibility to the disease and exposure to cancer-causing agents in the environment. Nevertheless, distinct attempts using molecular epidemiology have failed to demonstrate an interaction between susceptibility factors and such environmental factors. Thus, the solution does not seem to be so simple a relationship. I propose a model in which I integrate current theories concerning childhood ALL and, in addition, propose a new approach by which to study the causes of this infirmity.
In this model I propose that susceptibility to childhood ALL should not be viewed as being determined solely by a genetic factor or by whatever other individual factor. Rather, susceptibility should be understood as a phenomenon composed of different variables. It is these variables, working in conjunction, perhaps synergistically, which result in the phenomenon of susceptibility. That is, susceptibility to childhood ALL should not be seen as only one individual characteristic, rather, a cluster of interacting variables determines whether a child becomes susceptible to the disease.
More specifically, I propose the existence of a gradient of susceptibility in which an individual not only is, or is not, susceptible, but also may be found within a gradient, with the individual being more or less susceptible. (Note that the model does not exclude the existence of non-susceptible individuals). Similarly, such a susceptibility gradient may be composed of two principal components: 1) susceptibility to the disease, as is the case for some genetic rearrangements, such as MLL/AF4, ETV6/RUNX1, etc., and 2) susceptibility to the carcinogenic effect of environmental factors, as is the case for the genes related to methylation, which are involved in the development of ALL.
Similarly, exposure to environmental factors should not be understood only as either being exposed, or not exposed, to determined carcinogenic factor(s). Such exposure should also be regarded as a phenomenon that implies a gradient, ranging from no exposure, to little exposure, to high exposure to carcinogenic substances. This goes beyond considering only the exposure dose and implies instead exposure to several environmental factors or exposure to many carcinogenic factors. Exposures(s) could occur to the mother before conception or to the child during gestation and/or in the postnatal period before development of the disease. Consequently, according to this model, if there were exposure to many environmental carcinogens during one or more of these stages, the probability of the child developing ALL would be increased.
Taking into account both phenomena (susceptibility and exposure), we may then deduce that ALL is the result of the interaction between a gradient of susceptibility and a gradient of exposure to carcinogenic environmental factors. Thus, if an individual has high susceptibility to the disease, few environmental factors, or little exposure to such environmental factors, will be needed to develop ALL. Conversely, for the phenomenon of exposure, the greater the exposure that a child has to carcinogenic environmental factors, the less susceptibility to the disease would be needed to develop the illness (Fig. 1).
In addition to the foregoing, we consider it necessary to take into account another very important variable that has been considered in all the theories on ALL, especially in that for early pre-B lymphoblastic leukemia: the age of the child at the onset of the disease. In a manner analogous to that for other childhood cancers, it has been observed that the age of the child at onset of the cancer not only is a reflection of the degree of susceptibility to the disease (i.e., the most susceptible being the children in whom the cancer appears earliest, such as is the case for bilateral retinoblastoma or bilateral Wilms’ tumor, the age at onset of which is earlier than that of the unilateral forms), but also is a third phenomenon in the development of disease. We call this phenomenon the “vulnerability period” of a tissue and we define it as the stage of the greatest cellular proliferation of that tissue, which can be determined biologically either by developmental age or by provocation by external agents, e.g., infections.
This apparent period of vulnerability can be seen in other tumors; for example, for retinoblastoma. This vulnerable period appears primarily during the first three years of life, the stage of maturation of the retina; in osteosarcoma, the age at onset of the tumor is highly related with the growth spurt of the adolescent, in which there is a great cellular proliferation of bone tissue. Thus, with the incorporation of this vulnerable period as a third phenomenon in our model, the age at onset of the ALL reflects the degree of vulnerability of the tissues. Therefore, we conclude that ALL is the result of the interactions among the three phenomena described above: the gradient of susceptibility, the gradient of exposure to carcinogenic environmental factors, and the tissue vulnerability period (Fig. 2).
This model will permit investigators to redesign studies for identifying environmental risk factors related to ALL, as well as the studies that attempt to evaluate the importance of some characteristic related to susceptibility to ALL. In addition, the model encompasses the existing theories of some types of ALL, but above all, it permits the integration of knowledge about ALL to date, from the environmental aspects to the molecular processes involved in the onset of the disease. It is probable that this model may be modified for application to the majority of childhood cancers and, therefore, could change the manner in which investigators search for the cause of these infirmities.
Conflict of interestThe author declares no conflict of interest of any nature.