


In recent years, the use of high-throughput omics technologies has led to the rapid discovery of many candidate biomarkers. However, few of them have made the transition to the clinic. In this review, the promise of omics technologies to contribute to the process of biomarker development is described. An overview of the current state in this area is presented with examples of genomics, proteomics, transcriptomics, metabolomics and microbiomics biomarkers in the field of oncology, along with some proposed strategies to accelerate their validation and translation to improve the care of patients with neoplasms. The inherent complexity underlying neoplasms combined with the requirement of developing well-designed biomarker discovery processes based on omics technologies present a challenge for the effective development of biomarkers that may be useful in guiding therapies, addressing disease risks, and predicting clinical outcomes.
En los últimos años, el uso de las tecnologías ómicas de alta densidad de datos ha permitido el rápido descubrimiento de posibles biomarcadores. Sin embargo, esto no ha tenido un impacto notable en la clínica ya que se han implementado muy pocos de esos biomarcadores. En el presente documento se describe el potencial de las tecnologías ómicas en el desarrollo de nuevos biomarcadores. Con el objetivo de dar a conocer un panorama general de la situación actual, se comentan algunos ejemplos ilustrativos de biomarcadores genómicos, transcriptómicos, proteómicos, metabolómicos y microbiómicos en el campo de la investigación en oncología. Asimismo, se señalan algunas de las recomendaciones que se han propuesto para acelerar su validación e implementación, y se comenta sobre cómo la complejidad inherente a las enfermedades se combina con la complejidad de las tecnologías ómicas, de tal modo que el desarrollo de biomarcadores predictivos, pronósticos y diagnósticos eficientes plantea retos importantes.
Precision medicine, formerly known as personalized medicine, is a form of medicine that takes into account specific characteristics of a patient to individualize prevention, diagnosis, and treatment.1,2 Apart from the relevant clinical and epidemiological information, precision medicine relies on information provided by several omics fields that have sprung in the last decades: genomics, which studies the whole genome or a large subset of it (v. gr., the exome); transcriptomics, which deals with the full set of transcripts of a cell, tissue or organism; proteomics, comprising the study of the full set (or a large subset) of proteins present in a cell or tissue type; epigenomics, which investigates the complete set of covalent modifications of DNA that do not alter the DNA sequence itself but result in changes in gene activity; microbiomics, which concerns itself with the community of microbes and their genes in a patient; metabolomics, which analyses the complete set of low molecular weight metabolites (e. g., amino acids, organic acids, lipids, and sugars); and the field studying the exposome, which comprises molecules and events to which a person is exposed to (e. g., drugs, diet, and other environmental factors).3 Since the completion of the Human Genome Project, in 2003, the contribution of genomics to precision medicine has received the largest share of attention. The contribution of transcriptomics, proteomics, metabolomics, and other fields has been equally important.2,4
Omics technologies are high throughput techniques that make it possible to gather, in a single experiment, large amounts of data about a specific type of molecules, such as the three billion base pairs of the human genome, the universe of proteins in a given tissue or a large collection of metabolites. Examples of these technologies are next generation sequencing, used for genomics and transcriptomics studies, and mass spectrometry, used in proteomics and metabolomics studies.
The technological progress underpinning these omics technologies is bringing us closer to the realization of precision medicine. However, the contribution of omics technologies to precision medicine is not direct, but rather via the identification of relevant biomarkers. As defined by the World Health Organization, a biomarker is “any substance, structure or process that can be measured in the body or its products and influence or predict the incidence or outcome of the disease.”5 In a first step, omics technologies allow generating vast amounts of data on particular molecules (e. g., DNA, metabolites) in individuals with a specific condition.3,6,7 Data are then analyzed to determine whether particular biomarkers are associated with the occurrence of the disease or, perhaps, with a given prognosis, or even with a certain response to a defined therapeutic intervention. Upon identification, biomarkers are validated with other analytical platforms; usually, with those that are more likely to be found in a clinical laboratory, such as FISH, RT-PCR, PCR or immunoaffinity-based assays. At the end of several rounds of analytical and clinical validation, biomarkers may be approved to be used in the clinic.
The ability to produce a detailed characterization of a disease allows the stratification of patients into well-defined groups for tailored management and treatment, which are the basis of precision medicine.3,8,9
Biomarkers can be classified into four types: diagnostic biomarkers are used to determine the specific health disorder of the patient; prognostic biomarkers help to chart the likely course of the disease; predictive biomarkers indicate the probable response to a particular drug, and predisposition biomarkers indicate the risk of developing a disease.9,10
The successful identification of biomarkers has a long and distinguished history, which includes blood typing to guide blood transfusions,11 newborn metabolic screening for the early detection of metabolic diseases,12 analysis of serum prostate-specific antigen for the early detection of prostate cancer;13 overexpression/amplification of the HER2 receptor in breast cancer cells as a predictor of monoclonal antibodies response, like trastuzumab or pertuzumab;14 BCR-ABL translocation identification in chronic myeloid leukemia as a predictor of imatinib response,15 and HLA loci typing to reduce transplant rejection.16
The advent of high-throughput processes underpinning omics technologies is now contributing to the addition of novel biomarkers, some of which have already made the transition to the clinic.17 One such example is the expanded newborn metabolic screening by mass spectrometry.12 Another is the proliferation of disease-specific panels for the molecular diagnosis of genetic diseases using next-generation sequencing. In the field of oncology, the transcriptomics-derived biomarkers based on tumor gene expression profiling, Mamaprint, OncotypeDX, and PAM50, address the risk of recurrence in breast cancer.18–20 This type of gene expression profiling has also been used in the development of prognostic indexes for prostate and colon cancer,21,22 and hepatitis C-related early-stage cirrhosis.23 Genomics has also contributed to the development of the multitarget stool DNA test for detection of colorectal cancer.24
In this document, we present the various steps involved in biomarker development, followed by examples of biomarkers discovered using omics technologies that have successfully navigated the transition to the clinic in neoplasia.
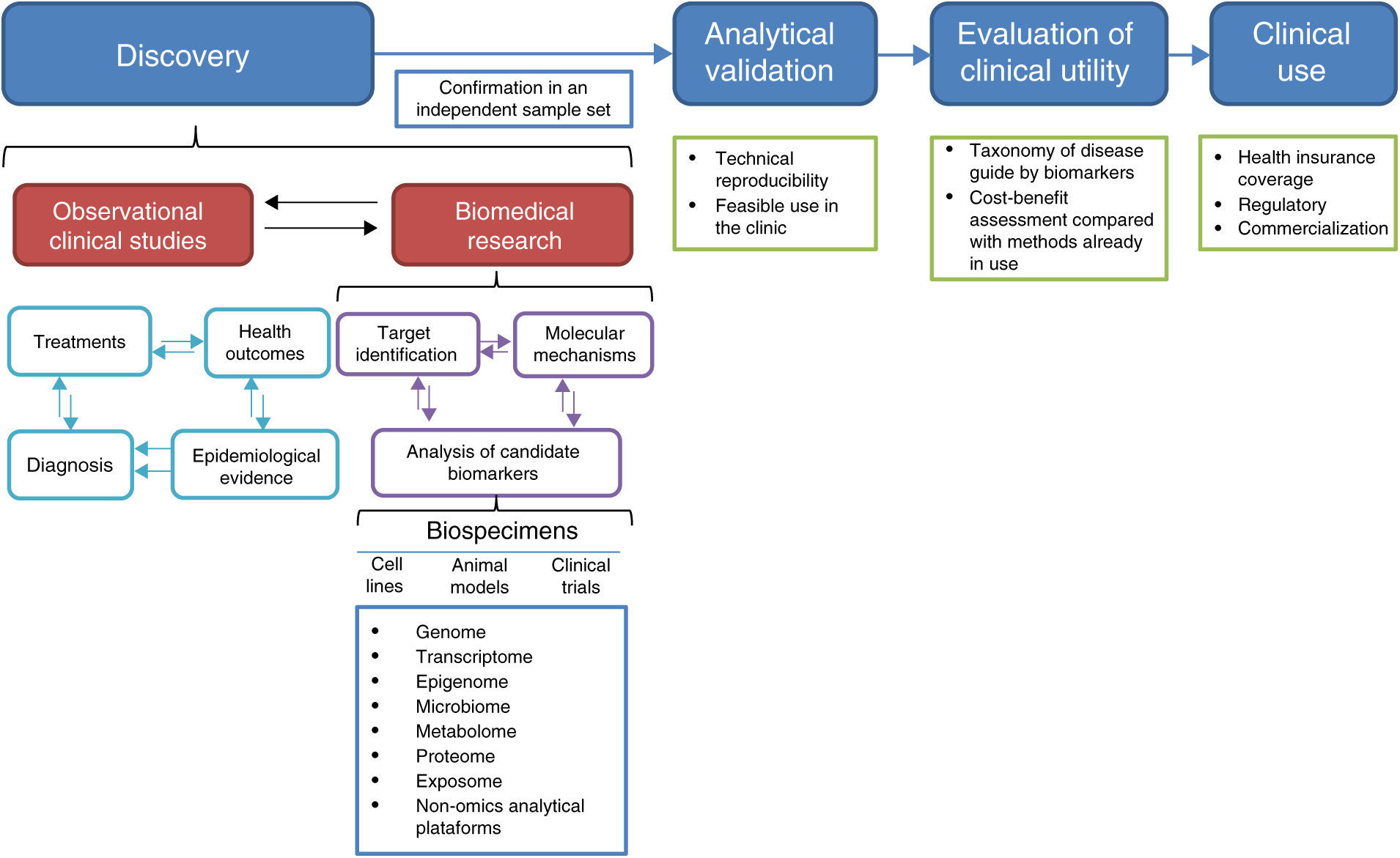
2Biomarker developmentThe process of biomarker development comprises four main steps: discovery, analytical validation, evaluation of clinical utility, and clinical use (Figure 1).25,26
2.1Discovery
In the discovery phase, the analysis of biospecimens leads to candidate biomarkers. Biospecimens may derive from cell lines, animal models, biopsies from existing cohorts, samples from patients enrolled in ongoing clinical trials, or archived samples from finished prospective studies or biobanks (Figure 1).3 Besides the identification of candidate biomarkers, this step may provide potential therapeutic targets and knowledge on the molecular mechanisms by which candidate biomarkers contribute to the pathological state.25 The information on these potential biomarkers must be interpreted in the context of all available information, such as the probable diagnosis, treatments undergone, epidemiological information about the disease, and health outcomes for the patients whose biospecimens were studied (Figure 1).
Ideally, specimens must be collected from large prospective case-control studies involving a clearly defined set of patients in a specific clinical context and as complete as possible information on the clinical characteristics, interventions and outcomes involved.10,25 Since it is not always possible to have such ideal conditions, which may also be costly and take a long time to achieve, many studies make use of archived specimens or biological models. At any rate, it is crucial to carefully define the inclusion criteria since poorly defined groups or heterogeneous samples may result in the development of signatures without therapeutic value.9 In this regard, it is worth mentioning that one of the main reasons why basic preclinical studies do not progress towards clinical applicability is that the samples used for biomarker discovery do not reflect the patient population in which those biomarkers are expected to be used.4,9 Another potential pitfall is sample heterogeneity that may result from deficiencies in the study design, such as non-matched confounding factors. Also, included in this category are poorly defined variables, such as the biospecimen source, the research question itself, target population, inclusion and exclusion criteria, and the endpoint of the study.9,25
Sample handling is also an important aspect to be considered in studies aimed at biomarker discovery. It is necessary to follow standardized protocols during sample collection, storage, and processing, as well as to use validated and well calibrated analytical methods to achieve robust and reproducible analyses.25 A crucial aspect during the discovery phase is the confirmation of the findings using an independent sample set.26
As stated before, the high throughput nature of the omics technologies is particularly well suited for biomarker discovery since it allows a detailed molecular characterization of biospecimens. However, poor reproducibility and the high number of false positives makes it necessary to undertake both analytical and clinical validation so as to confirm or reject the suitability of a candidate biomarker in diagnosing or predicting the disease of interest.25,26
2.2Analytical validationOnce promising biomarkers have been identified, it is necessary to assess their usefulness with the sort of tools normally available to a clinical laboratory, such as FISH, RT-PCR, PCR, HPLC or some immunoaffinity-based assay. Analytical validation of these tests must include dynamic range detection and reproducibility.25,26 If some of the complex omics technologies are to be used for routine clinical analysis, their technical reproducibility issues should also be addressed.3,27,28
The development of a combination of several types of molecules, as multilevel biomarkers, is an attractive option since the pathological state is determined by the complex interplay of various types of molecules, such as DNA, proteins, RNA and metabolites. However, analytical validation and determination of the statistical significance of such combinations require a higher number of studies than those necessary to develop a single-molecule biomarker.3,29 Currently, algorithms that integrate DNA methylation, copy number aberrations, point mutations and transcript levels in a multimodal signature are being developed, although there are some concerns about the size of the biopsy required to perform all the studies.29
2.3Evaluation of clinical utilityThe confirmation of the ability of a candidate biomarker to diagnose or predict the clinical outcome can be done in prospective clinical trials in which the biomarker may direct patient management, in prospective/retrospective studies analyzing archived specimens, or using samples from a biobank.25,26 At this stage, the studies aimed at prognostic biomarker evaluation may not necessarily influence clinical decision making. However, to increase the clinical utility of these studies, it has been recommended that the studies in which a companion therapeutic agent is evaluated, omics-based biomarkers should also be included.25
2.4Clinical useAfter the clinical usefulness has been demonstrated, the biomarker test must get regulatory approval, be commercialized, and incorporated into clinical practice guidelines.25,26
3Current state of omics-derived biomarker development in oncologyBiomarkers can positively impact patient care by predicting individual disease risk; allowing early detection of disease, which often increases treatment effectiveness; improving diagnostic classification, which in turn may promote individualized treatment; and monitoring the progress of a given therapy.9 However, very few omics-derived biomarkers have made their way to the oncology clinic so far.17,25 The complexity of cellular processes involved in tumor formation, heterogeneity of neoplasia (different tumors, intertumoral, and intratumoral), non-optimal study design, and poor methodological robustness and reproducibility are the main pitfalls that have contributed to the huge gap between the number of omics-based biomarkers found in basic research literature and those introduced to the clinic.9,17,29
Neoplasia is a particular type of disease. It may be seen as the outcome of proximate and ultimate causes. Among the latter are genetic and environmental factors that increase the risk of developing the disease (e. g., genetic variants in tumor-suppressor genes, smoking). These risk factors may lead to chromosome and genetic alterations that deregulate cell cycle and cell behavior. These alterations are the proximate (or actual) cause of the disease. Understanding the ultimate causes may lead to a better prevention and early detection, while the study of the proximate causes may allow the identification of biomarkers suitable for early detection, a more accurate prognosis and an individualized management of the patient, which is the basis of precision medicine. Omics technologies have been intensively used to study both the genetic risk factors and the mutation profiles associated with neoplasia. The approaches used for these two aspects differ. Genome-wide association studies (GWAS) are usually performed to identify genetic risk factors. These studies compare the distribution of common variants across the genome in a large set of cases and controls. In contrast, mutation profiles in tumors are studied through next generation sequencing (NGS) of the genome, the exome, the transcriptome, or a panel of genes known to be involved in a particular neoplasia. In an ideal scenario, tumor mutation profiling with NGS would involve more than one set of targets. For example, both tumor transcriptome and DNA sequence of a panel of genes might be studied in tandem.
GWAS are carried out to find cancer predisposition loci and have been the first choice approach for the last decade. However, the current diminishing cost of NGS has meant that the cost of the two approaches is converging; therefore, NGS might be used soon instead of GWAS in studies aimed at identifying genetic risk factors in neoplasia. NGS would have the advantage of providing information on the whole genome, as opposed to the 1-2 million sites currently interrogated by microarrays.
As stated above, SNPs across the genome of cases and controls are compared, and risk-associated loci are identified in GWAS.30 However, the proportion of individuals carrying a particular risk allele is usually low in the field of oncology.2,9,31 This situation has also been observed in other complex diseases—those resulting from the interplay of genetic risk variants and environmental factors as diabetes or arthritis. Some researchers think that the missing variation could be accounted for by including non-additive genetic effects, while others argue that rare variants, missed by GWAS, might underlie a larger number of cases than anticipated, in which case the common disease rare-variant scenario would be more important than it has been admitted so far.
Regarding the study of mutation profiles in tumors—that is, the actual cause of tumor phenotype—NGS has been the instrument of choice. Two of the most developed omics technologies are cancer genomics and transcriptomics. Tumor whole genome, whole exome, and transcriptome sequencing are now feasible assays with price tags on the order of 500 to 1500 USD,32,33 and comprehensive databases like TCGA,34 COSMIC35 or ICGC36 are publicly available. Cancer genomics has contributed significantly to the understanding of the biological basis of this disease.2,9 However, although still not widely used in the clinic,37 it has been making inroads, particularly in the most advanced hospitals around the world. One complicating factor when trying to identify biomarkers for precision medicine in oncology is that most tumors have a large number of alterations, sometimes running into thousands, but clearly not all of them are significant. The difficulty lies in distinguishing those that underlie the tumor phenotype (driver mutations) from those that result from the genomic and chromosomal instability associated with tumor formation, but which have no contributing effect to the tumoral phenotype (passenger mutations). For example, which mutations in a given gene deregulate protein function? Which proteins are important for cancer progression and chemoresistance? How an association of a genetic change with a specific treatment can be extended to different types of cancer?10
Another hurdle in developing precision medicine in neoplasia is the fact that there are far more numerous biomarkers than the drugs targeting the disrupted genes and proteins, irrespective of the number of useful biomarkers identified so far.2 There is still a long way to identifying most relevant neoplasia-associated biomarkers, which is a very active field where omics technologies are being used. These advances can be appreciated in the number of clinical trials that include biomarkers and omics-based biomarkers in their study design. A search on the clinical trials website of the U. S. National Institutes of Health (https://clinicaltrials.gov/) using the terms “biomarker,” “genomic marker,” “transcriptomic marker,” “proteomic marker” or “metabolomic marker” resulted in 20 290, 557, 31, 312 and 192 studies, respectively (accessed on February 21st 2017).
4Examples of biomarker discovery research in oncologyMany studies using omics technologies have been focused on cancer. The heterogeneous nature of the disease and the challenging events of relapse are ideal for biomarker-based patient stratification.3,25,38,39 In the following sections, selected reports, to exemplify the progress on biomarker discovery and how the various omics technologies can accelerate this process through the molecular characterization of cancer cells and their products, are commented.
4.1Pediatric acute lymphoblastic leukemiaTwo of the main challenges of cancer treatment are relapse and chemoresistance.40 In pediatric acute lymphoblastic leukemia (ALL), the use of omics technologies has led to the identification of several signatures that predict a higher probability of relapse, as well as identifying some common pathways that malignant blasts use to evade therapy and should be considered in the exploration of therapeutic targets.38 These signatures include up-regulation of genes involved in proliferation, cell cycle regulation and apoptosis (BIRC5, FOXM1, GTSE1, DUSP6, F2R, HRK), DNA repair (FANCD2, PTTG1, UBE2V1), drug resistance (RAB5C), nucleotide biosynthesis (TYMS, CAD, PAICS, ATIC, DHFR), cellular differentiation (HMGA1), and deregulation of the glucocorticoid, WNT and MAPK signaling pathways.38 Transcriptomics signatures with predictive value41,42 led to the discovery of epigenetic reprogramming as a means to restore chemosensitivity in B-lineage leukemia cell lines and primary B-lymphoblastic leukemia patient samples in preclinical studies. This reprogramming made use of the histone deacetylase inhibitor vorinostat in combination with the DNA methyltransferase inhibitor decitabine (1μM each).43 The results of the subsequent clinical trial (NCT01483690) have not been published yet. Prognostic biomarkers have also been identified by genomics44–47 and proteomics48–52 studies. An example of how a proteomics study has gone deeper into a mechanistic knowledge that identifies therapeutic targets is the study of Nicholson et al.50. The quantitative proteomic comparison of nuclear lysates from the glucocorticoid sensitive B-ALL cell line PreB67 and the resistant subline R3F9, indicated that reduced expression of the glucocorticoid receptor target genes and differentiation from preB-II to an immature B-lymphocyte stage fostered dexamethasone resistance. This effect was associated with the activation of the JNK signaling pathway to the extent that 5μM of the JNK inhibitor SP600125 reduced 30-fold dexamethasone tolerance of the resistant cell line.50 Dehghan-Nayeri et al. identified three potential dexamethasone-resistant prognostic biomarkers: voltage-dependent anion channel 1 (VDAC1), sorting nexin 3 (SNX3) and pre-folding subunit 6 (PFDN6).52 Using the dexamethasone-resistant B-ALL cell line REH and bone marrow from patients with standard risk (n=10), high risk (n=7) and a control group (n=7), the authors concluded that the reduced expression of these proteins was associated with dexamethasone resistance.52 Cellular roles of VDAC1 include drug resistance and regulation of apoptosis, whereas SNX3 is involved in protein trafficking and PFDN6 in protein folding.52 In a similar study, Jiang et al. identified the proliferating cell nuclear antigen (PCNA), which is involved in cell cycle regulation and survival, as a candidate prognostic marker of prednisolone response in ALL.49 Using cell lines and bone marrow from 43 patients, the authors concluded that the reduced expression of PCNA after eight days of treatment represents a promising prognostic biomarker of good response.49 Interestingly, the use of prednisolone did not reduce the expression of VDAC1 in the REH cell line;49 however, dexamethasone did,52 suggesting that changes in protein expression could be glucocorticoid-specific.
Quantitative proteomics analyses can also contribute to refining stratification of patients with ALL, as demonstrated by Xu et al.51 In this study, a quantitative comparison of the bone marrow proteome of 12 newly diagnosed patients with B-ALL was performed. Patients were classified into two groups: the low/medium- and the high-risk groups (six patients each), plus a control group (six patients as well) with non-malignant hematological disorders. They found 86 differentially expressed proteins that may be used to stratify patients more accurately. The relevant proteins were involved in pre-mRNA splicing, DNA damage response and stress response. Five proteins were selected for validation in bone marrow from an additional 24 low/medium-, and 18 high-risk patients. Although statistically significant, the increased expression of Hsp90β, Hsp90α, YBX1, DDX48 and Thrp3 showed high variability for which this may preclude their use as single-molecule biomarkers. Further validation of these results is needed, and probably the combination of data with other prognostic risk factors might yield an increased statistical power for the resultant multilevel biomarker test.3
Ideally, biomarkers should be analyzed in non-invasive biospecimens like blood, plasma, serum, urine, saliva or stool. In the case of ALL, analysis of blood is less invasive than bone marrow. In this regard, Cavalcante et al.48 analyzed the glycosylated proteome in serum from ten patients and identified nine proteins present at early stages, but not during remission nor in the control group. The authors proposed that this panel of candidate protein biomarkers might improve early diagnosis. Omics-derived biomarkers in ALL are still not helping pediatric oncologists in decision making. However, the detailed molecular characterization of biospecimens contributes to the knowledge of the underlying molecular mechanisms of the disease, a requisite for the development of further large-scale prospective studies in which the high-throughput analyses will accompany the study of treatment response in representative cohorts of specific subgroups of patients.7,25,29
4.2Solid tumorsNon-invasive biomarkers have also been reported for detection and monitoring solid tumors. For example, Bettegowda et al. quantified circulating tumor DNA (ctDNA) for the detection of pancreatic, ovarian, colorectal, bladder, melanoma, gastroesophageal, breast, hepatocellular, head and neck cancer.53 In 5ml of plasma, these authors detected ctDNA in 75% of the patients (640), and the concentration of ctDNA correlated inversely with survival rates. Moreover, ctDNA was detected in 82% of patients with metastatic cancer, in contrast to 55% of patients without metastasis. NGS was used for exome or whole genome sequencing of biopsies from solid tumors to identify mutations in each patient. Although the initial genetic profiling of the tumor was necessary to track ctDNA, the proposed strategy allowed the detection of known clinically relevant K-Ras mutations in metastatic colorectal cancer patients with a sensitivity of 87.2% (the test was positive for 87.2% of patients) and a specificity of 99.2% (the test was negative in 99.2% of healthy volunteers used as a control group) in tests with no previous solid tumor genetic profiling.53
Metabolites in liquid biopsies like serum or urine may reflect the biochemical state of the patient, which is the result of the complex interactions among drugs, gene expression, proteins, age, microbiota, environment, and disease. Thus, metabolomic profiles are promising sources for non-invasive biomarkers. Miolo et al. compared the plasma metabolome from 34 patients with HER-2 positive breast cancer with different responses to the trastuzumab-paclitaxel neoadjuvant therapy.54 The metabolomics profile before treatment showed that the patients with a high concentration of spermidine and low concentration of tryptophan had a higher probability of achieving a complete response, compared to patients with the opposite trend in the balance of these two metabolites. The tryptophan/spermidine ratio was used to establish a threshold value and to construct a receiving operating characteristic curve (ROC), which resulted in a predictive power with a sensitivity of 90% and a specificity of 87%. Regarding urinary metabolomics, Jin et al. compared the urine profiles from 138 patients with bladder cancer, 69 healthy volunteers and 52 patients with hematuria due to non-malignant diseases.55 These authors identified 12 metabolites that allowed differentiation of patients with bladder cancer from the other two groups. The diagnostic performance of these findings, evaluated with a ROC curve, resulted in a sensitivity and specificity of 85%.
Another example of non-invasive biomarker discovery is the work of Kim et al. who performed a proteomics study in plasma to develop a promising biomarker for early detection of non-small cell lung cancer (NSCLC).56 As early detection of cancer increases the likelihood of a good outcome, the authors proposed a targeted proteomics pipeline for verification of blood-based biomarkers. They selected and analyzed 95 proteins in plasma from 72 patients with NSCLC (of different types and stages) and 30 healthy volunteers. This investigation led to the discovery of the protein zyxin, a cell-adhesion, and mechanotransducer protein, as a potential biomarker for the early detection of NSCLC.
Besides blood and urine, stool may also be a good source for non-invasive biomarkers. Kostic et al. performed whole genome sequencing analysis of the colorectal cancer microbiome, comparing 104 matched tumors versus healthy tissue biopsies. Besides an altered microbiota, the authors found a significant enrichment of Fusobacterium species in the tumor microenvironment.57 Furthermore, in a later study, the same group demonstrated that Fusobacterium nucleatum promoted tumor progression in a mouse model of colon cancer which was accompanied by an increase in infiltrating tumor-permissive myeloid-derived suppressor cells and by a proinflammatory expression signature.58 These results suggested that by reducing the number of Fusobacterium species in the intestinal tract, tumor progression in colorectal cancer patients could be delayed.58
MicroRNAs (miRNAs) are small non-coding RNA molecules that can bind to mRNA, thereby reducing the rate of translation.59 As negative regulators of gene expression, some miRNAs have been proposed as biomarkers in cancer.59 Simmer et al. discovered that the low expression of the miR-143 in primary tumors from 55 patients with colorectal cancer correlated with an increased median progression free survival in response to a capecitabine-based treatment.60
All the above mentioned biomedical studies share the aim of developing markers with clinical utility. However, clearly, some studies have reached more progress than others in this respect. The recommendations to accelerate the translation of the findings from biomedical research to the clinics have been reviewed.7,9,25,61
5Concluding remarksThe advent of the omics technologies has boosted the ability to characterize biospecimens at the molecular level. In the years to come, high-throughput analyses are expected to co-evolve with biomarker-based precision medicine leading to better patient care. The complexity of the pathological state poses enormous challenges, and the various omics technologies still have technical issues like reproducibility and a high false positive rate. However, the joint effort of clinicians, researchers, bioinformaticians, and biostatisticians, in academia and industry will certainly make progress towards the development of sensitive and specific predictive, prognostic and diagnostic biomarkers.
The high-throughput nature of the omics technologies is enabling the fast discovery of candidate biomarkers with the results being described in a large number of preclinical reports. Much slower and time-consuming, large prospective well-designed studies will be essential for clinical validation. Deep mechanistic studies are also necessary for the development of viable companion therapeutics. Reduced costs and increased reproducibility will benefit the biomarker development process and may contribute to the continuous reevaluation of the classification of patients.
FundingFederal funding: HIM/2015/053 SSA1183 and HIM/2016/026 SSA1222. CONACYT, grant 589666 (for AGO).
Conflict of interestThe authors declare no conflicts of interest of any nature.