





Iron is essential for cell growth and is imported into cells in part through the action of transferrin (Tf), a protein that binds its receptor (TfR1 or CD71) on the surface of a cell, and then releases iron into endosomes. TfR1 is a single pass type-II transmembrane protein expressed at basal levels in most tissues. High expression of TfR1 is typically associated with rapidly proliferating cells, including various types of cancer. TfR1 is targeted by experimental therapeutics for several reasons: its cell surface accessibility, constitutive endocytosis into cells, essential role in cell growth and proliferation, and its overexpression by cancer cells. Among the therapeutic agents used to target TfR1, antibodies stand out due to their remarkable specificity and affinity. Clinical trials are being conducted to evaluate the safety and efficacy of agents targeting TfR1 in cancer patients with promising results. These observations suggest that therapies targeting TfR1 as direct therapeutics or delivery conduits remain an attractive alternative for the treatment of cancers that overexpress the receptor.
El hierro es esencial para el crecimiento celular. Es transportado dentro de las células con la ayuda de la transferrina (Tf), proteína que se une a su receptor (TfR1 o CD71) en la superficie celular y libera el hierro dentro de los endosomas. El TfR1 es una proteína de membrana tipo II que se sobreexpresa en muchos tejidos debido al requerimiento de las células para importar hierro unido a Tf. La sobreexpresión de TfR1 se ha asociado con células que proliferan rápidamente, incluyendo los diferentes tipos de cáncer. El TfR1 se ha empleado como blanco terapéutico por diversos motivos: su accesibilidad a la superficie celular, su capacidad de internalizarse constitutivamente en las células, su papel esencial en el crecimiento y la proliferación celular, así como por su sobreexpresión en las células tumorales proliferantes. Entre los agentes terapéuticos dirigidos contra el TfR1 destacan los anticuerpos, por su alta especificidad, estabilidad y propiedades estructurales. Se han realizado diversos ensayos clínicos para evaluar la seguridad y la eficacia de los anticuerpos que reconocen el TfR1 en pacientes con cáncer y se han obtenido resultados prometedores. Estas observaciones sugieren que las terapias con fundamento en el reconocimiento de TfR1, ya sea como terapia directa o empleados como acarreadores, representan una alternativa muy atractiva de tratamiento contra los diferentes tipos de cáncer que sobreexpresan este receptor.
As an essential cofactor for life, iron facilitates several enzymatic reactions critical for DNA replication and cellular respiration. The cellular processes that rely on iron include nucleotide synthesis and electron transport. However, free ferrous iron can be toxic to living systems.1 To avoid this toxicity, organisms have evolved iron transport and storage systems such as carrier proteins (transferrin and ferritin), heme, and iron-sulfur clusters.1 Transferrin (Tf) is found in the blood of mammals as a two-lobed protein.2 Under neutral physiological conditions, each transferrin molecule is capable of binding two ferric iron atoms. Tf releases its iron cargo at low pH, with maximal release observed near pH 5.3 Definitive evidence of a transferrin receptor (TfR1) was found first in rabbit reticulocytes,4 and later confirmed in a variety of species and cell types, including human placenta, a rich source of TfR1.5,6 TfR1 is a cell surface protein that binds Tf and facilitates its endocytosis from plasma.2 The receptor exists on the cell surface as a homodimeric type-II glycoprotein receptor; the extracellular domain (ECD) of the homodimer can bind up to two molecules of transferrin. The ECD of TfR1 contains three subdomains: a helical, protease-like, and apical domain (Figure 1).
, helical (H, green) and protease-like domain (P, blue).")
Scheme of the TfR1 homodimer on the cell surface consisting of two monomers linked by disulfide bridges at cysteines 89 and 98. The TfR1 contains an intracellular domain, a transmembrane domain, and a large extracellular domain. Here, the structural model of the extracellular domain of TfR1 was generated with coordinates from PDB ID 1DE4, and consists of three subdomains: apical (A, orange), helical (H, green) and protease-like domain (P, blue).
TfR1 is encoded by the TfRC gene that belongs to the transferrin receptor family. The family of receptors includes TfR2 and is derived from ancient carboxypeptidases.7 The TfR1 homodimer is held together by disulfide linkages and constitutively endocytosed through the canonical clathrin-mediated pathway. Once in acidified endosomes, a receptor carrying iron loaded Tf undergoes a structural rearrangement that promotes the release of iron by Tf. Meanwhile, the iron-free Tf molecule remains bound to the receptor and recycles back to the cell surface where it is released at a neutral pH (Figure 2A). Recycling of TfR can occur hundreds of times during the lifetime of a single receptor.8 At any given time, a cell can express hundreds of thousands of copies of TfR19 with only a small percentage present at the cell surface. More detailed descriptions of Tf trafficking and iron import are available elsewhere.10,11
 Endocytosis of iron loaded Tf bound to TfR1 via clathrin-coated pits into the endosome compartment. Acidification through proton pumping into the endosome induces a conformational modification in Tf that results in the release of iron. The iron is then pumped into the cytosol from the endosome by the divalent metal transporter 1 (DMT1). Tf is later released once the TfR1 reaches the cell surface again. (B) On the left, the TfR1 binding to Tf (red) and on the right side, binding to the Hemochromatosis protein (HFE, blue), a natural ligand of the receptor, modeled using coordinates from PDB IDs 1DE4 and 3S9L.")
Iron internalization and TfR1 ligands. (A) Endocytosis of iron loaded Tf bound to TfR1 via clathrin-coated pits into the endosome compartment. Acidification through proton pumping into the endosome induces a conformational modification in Tf that results in the release of iron. The iron is then pumped into the cytosol from the endosome by the divalent metal transporter 1 (DMT1). Tf is later released once the TfR1 reaches the cell surface again. (B) On the left, the TfR1 binding to Tf (red) and on the right side, binding to the Hemochromatosis protein (HFE, blue), a natural ligand of the receptor, modeled using coordinates from PDB IDs 1DE4 and 3S9L.
All three subdomains of TfR1 are required for Tf binding. The helical domain of TfR1 is responsible for dimerization. The protease-like domain resembles the glutamate carboxypeptidase family of membrane-associated proteases, which include the prostate-specific membrane antigen (PSMA).7 TfR1 lacks protease activity due to evolutionary changes in key residues within its ancestral active site. TfR1 is degraded in the lysosome and is dependent on a low pH environment.12 When shedding from the membrane, the extracellular domain of TfR1 can be found in the interstitial spaces and circulating in the blood, where it has become a diagnostic marker of iron homeostasis.13 A second receptor, TfR2, is expressed most highly in the liver.14 but also in cancers including glioblastoma.15
2Structure of TfR1 alone and in complex with its natural ligandsIn 1999, the structure of the extracellular domain (ECD) of human TfR1 was first determined by crystallographic means to 3.2Å resolution (Figure 1).16 The overall fold of each TfR chain resembles that of carboxy or aminopeptidases. The structure also revealed the first N-acetylglucosamine residue at each of three asparagine-linked glycosylation sites.
The protease-like domain of the TfR1 structure contains a central beta sheet composed of seven strands and flanking helices.17 Strand 6 within the central beta sheet of the protease-like domain shows an unusual disulfide bridge, spanning a single residue. The protease-like domain is linked to the protease-associated domain by a pair of beta strands.17 The protease-associated domain resembles a beta sandwich that fits as an insert into the sequence of the protease-like domain. This may have been the evolutionary product of a gene transplant.7 In close contact with both the protease and apical domains is the helical domain of the receptor, comprised predominantly of a pair of parallel alpha-hairpins.16 The ectodomain of the receptor sits about 30Å above the membrane and is linked to a single pass transmembrane region near its protease-like domain. This linker is the site of disulfide bridges above the membrane (Figure 1).
The molecular details of the interaction between TfR1 and both its ligands, Tf and human hemochromatosis protein (HFE), have been resolved with crystallographic detail (Figure 2; PDB ID 1DE4 and 3S9L). The structure of HFE bound to TfR1 reveals a one to one stoichiometry of HFE to TfR1.18 Each HFE molecule binds TfR1 in plane, meaning HFE is likely to bind only TfR1 present on the membrane of the same cell. A three-helix bundle predominantly stabilizes the complex of HFE and TfR1. This bundle is formed by one helix from HFE and two from the helical domain of TfR1.17 While HFE remains relatively unchanged by the interaction with TfR1, the helical domain of the receptor undergoes a slight rearrangement upon binding.
The structure of Tf bound to TfR1 was solved by crystallographic means at room temperature and pH 7.5.18 Iron-loaded human Tf bound to its receptor reveals restructuring of the TfR1 dimer interface. In this structure, two accompanying Tf molecules bind the TfR1 homodimer, with both the N and C lobes of Tf demonstrating significant interactions with the receptor.18 On TfR1, binding to Tf generates a histidine cluster that is primed for conformational rearrangement upon exposure to low pH.19
The molecular details underpinning the interactions of TfR1 with its natural ligands are the foundation upon which targeting studies must rely for accurate and specific targeting of the receptor. Therapeutics that are derivatives of the natural ligands immediately benefit from the knowledge of these interactions. Importantly, the knowledge of the atomic structures of these complexes allows for the molecular design of proteins or small molecules19 that can modulate the activity of TfR1 and thus serve as direct anticancer therapeutics or enhance the function of other anticancer agents.
3Expression of TfR1 in malignant cellsAnalysis of malignant cells from various tissue origins20,21 has revealed a positive correlation between receptor expression and cell proliferation.22 This is not surprising given that malignant cells have an intrinsically high demand for iron as a cofactor for DNA synthesis, and the iron-mediated regulation of molecules that control cell cycle progression.23 Altogether, it is clear that an abundance of TfR1 is essential for the survival of certain tumor cells. This is particularly true for hematopoietic malignancies, given the central role played by TfR1 in the development and function of the adaptive immune system.24 In fact, patients that show combined immunodeficiency with impaired B and T cell function that is caused by a homozygous mutation of tyrosine 20 to histidine in TfR1 have been identified.24 A large cohort of patients with chronic lymphocytic leukemia express both TfR1 and TfR2 at high levels, perhaps regulated by post-transcriptional control of its expression,25 and both TfR1 and TfR2 are expressed at high levels in erythroleukemia but not in acute myeloid leukemia.26 TfR1 is reported to also be overexpressed in certain solid tumors.8
4Targeting TfR1TfR1 is a widely accessible portal into a variety of cells overexpressing the receptor. Since the mode of entry of TfR1 into cells is constitutive, and the molecular underpinnings of its interactions with ligands are well known, both can be exploited by some targeting strategies.8,27 Different approaches have been employed to target TfR1 for direct inhibition of cell proliferation or the delivery of agents into cells.9 However, we will focus on the strategies that include the use of antibodies. Antibodies used as therapeutics to target TfR1 usually bind its ECD and perform one of two tasks: block binding of Tf to the receptor and delivery of cargo into cells. These strategies include but are not limited to antibodies and fragments of antibodies alone or combined with small molecules, proteins, nucleic acids, and nanoparticles (Figure 3).8

Antibody strategies to target TfR1 in malignant cells. The therapeutic approaches include monoclonal antibodies alone or combined with therapeutic agents, such as small molecules chemotherapeutic drugs, protein toxins or genes in vectors enclosed in nanocarriers. Targeting can be achieved by whole antibodies or single chain antibody fragments specific for the extracellular domain of the TfR1.
Due to their exquisite specificity and high affinity, a variety of antibodies have been designed to target TfR1.28 The first monoclonal antibody to specifically target TfR1 was used in an attempt to curtail tumor cell growth: a murine IgG1 named B3/25 that antagonizes Tf binding to the receptor.29 Since then, antibodies have been developed to bind a number of different epitopes on TfR1; most antibodies were designed for either delivery purposes, or to inhibit receptor function directly.27 Studies have measured the effect of antibodies on TfR1 expression, recycling, and ability to facilitate Tf-mediated iron uptake in cells.30–34 Antagonistic antibodies or antibody fragments prevent the binding of Tf directly to TfR1 and can starve cells of Tf-bound iron by competition or steric hindrance. An example of an antagonistic antibody targeting TfR1 with high affinity is A24: it competes with Tf for receptor binding, and induces TfR1 degradation, leading to intracellular iron starvation in adult T-cell leukemia cells in vitro and in vivo.35 Other examples of antagonistic anti-TfR1 antibodies against cancer with different configuration include scFvs with human variable domains generated by phage display that alone in bivalent format (F12CH and H7CH), could block cell proliferation in vitro and mouse models of erythroleukemia.36
Non-antagonistic antibodies can bind and affect TfR1 but do not competitively inhibit Tf uptake and are therefore less toxic. A cohort of thirty-two mouse antibodies was generated in the late 1980s for targeting of TfR1. These antibodies were all generated with specificity for the extracellular domain of TfR1. Few of these were capable of significantly blocking the proliferation of cells expressing high levels of TfR1.32 The others failed to block either binding or internalization of Tf to TfR1. The variable region of one of these antibodies, 128.1, would go on to be used as a vehicle for TfR1-mediated delivery into the brain or malignant tissues.33,37,38 Based on the variable region of 128.1, a fusion protein was developed comprised of a mouse/human chimeric antibody (ch128.1, previously known as anti-hTfR1 IgG3) with chicken avidin (Av).33
The original intent of constructing ch128.1Av was to target TfR1 as a universal delivery vehicle for biotinylated agents into cells expressing high levels of TfR1.33 However, the unexpected discovery that ch128.1Av and a mouse/human chimeric anti-rat TfR IgG3-Av fusion protein were both intrinsically cytotoxic to malignant hematopoietic cells, prompted the further investigation of the nature of the interaction between these non-neutralizing antibodies and TfR1.33,38 The ch128.1Av antibody proved highly cytotoxic to malignant B cells, and primary cells isolated from MM patients.34 The fusion protein is also toxic to lymphoma and leukemia cell lines. Thus, fusing avidin to this anti-TfR1 Ab enhances its cytotoxicity resulting in TfR1 sequestration and degradation, and substantial disruption of iron homeostasis, leading to cell death.39
6Delivery of anticancer agents through antibodies targeting TfR1The high specificity of antibodies against TfR1 provides an optimal tool to facilitate the accumulation of non-selective therapeutic agents at the tumor site, increase the intracellular concentration, improve anticancer activity, reduce exposure of healthy cells, and improve the overall therapeutic efficacy. Multiple delivery strategies using antibodies or fragments of variable region held together with a flexible linker (scFv) targeting TfR1 have been developed against cancer cells (Figure 3). Antibodies can deliver a variety of agents by the direct linkage to the therapeutic moiety or by linkage to carriers loaded with a therapeutic cargo.
Antibodies directly conjugated to chemotherapeutic drugs, also known as immunoconjugates, have shown anticancer efficacy targeting TfR1. The murine IgG1 anti-human TfR (5E9) conjugated to doxorubicin (ADR) with a pH sensitive linker have demonstrated the relevance of the immunoconjugate internalization via TfR for efficacy against human hematopoietic malignant cell lines Daudi and Raji in vitro and in vivo.40
The combination of monoclonal antibodies with radioisotopes, known as radioimmunotherapy, has also been used in TfR1 targeted therapies. Recently, a completely human anti-TfR1 conjugated to the β-emitting radionuclide 90Y has been generated. This conjugated antibody allowed the accumulation of the radioisotope in a xenograft model of pancreatic tumor cells overexpressing TfR1 but not in normal organs, preventing the tumor growth.41
Some toxic compounds have been coupled to full IgG antibodies to generate immunotoxins for cancer therapy. Plant toxins, such as ricin42 and saporin,43–45 or bacterial toxins including Pseudomonas exotoxin (PE),46 diphtheria toxin (DE),47 the fungal toxin alpha-sarcin obtained from Aspergillus giganteus, which interacts with the ribosome and inhibits protein synthesis, and even enzymes such as bovine pancreatic ribonuclease A have been complexed to anti-TfR antibodies, and tested against different malignant conditions, including leptomeningeal neoplasia, B-cell lymphoma, epidermoid tumor, and glioblastoma, showing a promising response. Since the targeting domain is the most relevant for immunotoxins, scFv antibody fragments specific for TfR1 have also been combined with peptide toxins. A scFv of the anti-TfR1 HB21 has been genetically fused to the truncated mutant of Pseudomonas exotoxin (PE40) exhibiting cytotoxicity against colon carcinoma expressing high levels of TfR1 in vitro and in vivo, showing efficacy also against human epidermoid carcinoma, prostate carcinoma, ovarian carcinoma, and breast carcinoma.48
The use of carriers combined to anti-TfR1 antibodies allows the intracellular delivery of different agents into cancer cells that can be covalently bound, entrapped, or adsorbed to the particle, avoiding the immune system inactivation, and large size or solubility issues. Particles such as liposomes carrying small molecules or nucleic acids can be conjugated to whole antibodies or scFv forming immunoliposomes. These systems targeted against TfR1 can actively deliver their cargo into tumors, enhancing the therapeutic activity of the anticancer compounds. The anti-TfR1 antibody OKT9 has been conjugated to liposomes loaded with ADR that showed significant cytotoxicity against the ADR-resistant human leukemia cell line K562/ADM.49 Liposomes complexed to the scFv anti-TfR1 5E9 have been used as a low toxicity, systemic gene delivery system that selectively targets tumor cells systemically delivering the p53 tumor suppressor gene.50 These immunoliposomes showed improved targeted gene delivery and transfection efficiency of p53 gene in vitro and in vivo in a human breast cancer metastasis model that–in combination with docetaxel– resulted in prolonged survival with improved efficacy. These studies suggest that targeting cancer cells through TfR1 is a promising approach to the treatment of malignant conditions overexpressing this membrane protein.
7Targeting TfR1 in a clinical settingFor a targeted therapy to be successful, it must exert strong action predominantly on the target cell while sparing the bystander cells and minimizing unwanted side effects. Antibodies are particularly equipped to do so given their high specificity and affinity. However, a protein that is ubiquitously expressed, such as TfR1, poses a challenge for targeted therapies. Therapy must overcome the ubiquitous expression by exploiting a therapeutic window, a difference between the level or type of expression in malignant cells compared to their normal counterparts.
Although TfR1 is ubiquitously expressed, its particularly high expression in rapidly proliferating cells, including hematologic malignancies, defines it as a viable molecular target. The targeting of TfR1 has already proven to be more efficient than other pleiotropic treatment strategies including blunt chemotherapeutic agents.34,51 In addition, the capacity to target TfR1 makes it a useful tool for molecular targeting and delivery of toxic agents into malignant cells.
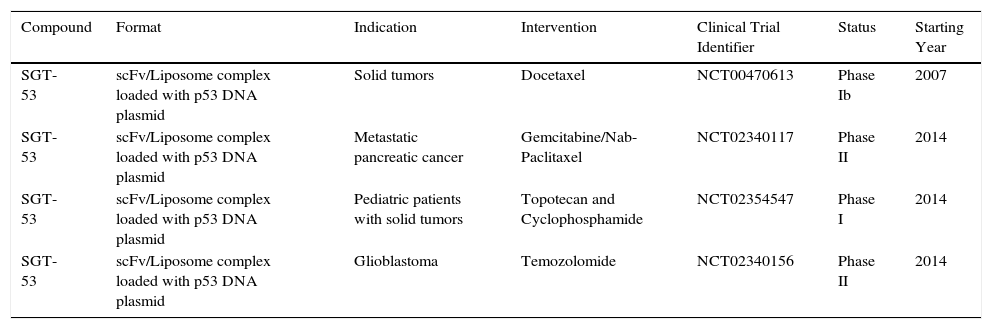
While a therapeutic agent targeting TfR1 for cancer therapy has yet to obtain FDA-approval, various efforts are underway to improve the specificity and efficacy of molecules that target the receptor in clinical and pre-clinical settings. Clinical efforts are underway focused on the evaluation of SGT-53, a scFv anti-TfR1 liposome complex loaded with p53 DNA for gene therapy (Table 1). A Phase Ib dose-escalation clinical trial was performed to assess the combination of SGT-53 and docetaxel in 14 patients with advanced solid tumors (NCT00470613).52 The combination was well tolerated and exhibited clinical activity: three patients achieved partial response with tumor shrinkage of -47%, -51% and -79% and two other patients had a stable disease with significant tumor reduction of -25% and -16%. The positive results obtained warrant further evaluation in phase II trials.
Open Clinical Trials of therapeutic agents against cancer targeting TfR1.1.
| Compound | Format | Indication | Intervention | Clinical Trial Identifier | Status | Starting Year |
|---|---|---|---|---|---|---|
| SGT-53 | scFv/Liposome complex loaded with p53 DNA plasmid | Solid tumors | Docetaxel | NCT00470613 | Phase Ib | 2007 |
| SGT-53 | scFv/Liposome complex loaded with p53 DNA plasmid | Metastatic pancreatic cancer | Gemcitabine/Nab-Paclitaxel | NCT02340117 | Phase II | 2014 |
| SGT-53 | scFv/Liposome complex loaded with p53 DNA plasmid | Pediatric patients with solid tumors | Topotecan and Cyclophosphamide | NCT02354547 | Phase I | 2014 |
| SGT-53 | scFv/Liposome complex loaded with p53 DNA plasmid | Glioblastoma | Temozolomide | NCT02340156 | Phase II | 2014 |
TfR1 is an attractive target for therapeutic strategies aiming to curb the growth of cancer cells. Cancer is one of the leading causes of death in the world, despite worldwide efforts to limit its devastating effects and find a cure. New tools for fighting hematologic malignancies still require a better understanding of the mechanisms by which current and developing therapies succeed and fail. We have outlined a brief history of the emergence of TfR1 as a target for the treatment of malignancies and summarize efforts to target this receptor by various promising therapeutics. Future efforts in tumor targeting will benefit from an improved understanding of the molecular underpinnings of TfR1 function.
FundingThe authors are supported by CONICET PIP (114-2011-01-00139), Fundación Bunge y Born, and Fundación René Barón. G.H. is a member of the National Council for Scientific and Technological Research (CONICET), Argentina. R.L.P. acknowledges support from CONACYT (CB-2013-01-222446, INFR-2015-01- 255341) and Fondos Federales (HIM/2013/022 SSA.1067, HIM/2015/002 SSA.1189).
Conflict of interestThe authors declare no conflicts of interest of any nature.