


Paciente masculino de 4 años 11 meses de edad, quien fue referido de un hospital de segundo nivel por aumento de volumen de ganglios submandibulares, axilares e inguinales de dos meses de evolución, acompañados de dolor inguinal y con la presencia de fiebre de 39°C.
1.1Antecedentes no patológicosOriginario y residente de Zumpango, Estado de México, de nivel socioeconómico bajo.
1.2Antecedentes perinatales y patológicosProducto de gesta I, control prenatal regular, amenaza de parto pretérmino a las 24 SDG, que se manejó con reposo. Nació de término vía abdominal por circular de cordón de cuello, pesó 3,300g, midió 51cm. Egresó sin complicaciones.
1.3Padecimiento actual07-23 noviembre 11. A los 4 años 4 meses de edad es referido al Hospital Infantil de México Federico Gómez (HIMFG) por la presencia de adenomegalias. El abordaje inicial es por consulta externa (CE) de Cirugía General con biopsia de ganglio cervical izquierdo. Se encontró conglomerado ganglionar compatible con enfermedad de Castleman (variedad hialinovascular) por medio de tinciones de inmunohistoquímica (CD10, CD20, CD3 y BCL-6, positivas; LMP-1 y CD30, negativas) y aspirado de médula ósea negativo a infiltración neoplásica.
26-31 diciembre 11. Se realizó linfadenectomía cervical bilateral radical con resultado histopatológico de enfermedad de Castleman variedad hialinovascular.
16 febrero-26 marzo 12. Ingresó a Urgencias por padecimiento de 15 días de evolución con disfagia de intensidad moderada, placas blanquecinas en cavidad bucal que se desprendían fácilmente, dolorosas, además de tos productiva, cianozante y disneizante de 24h de evolución, acompañada de fiebre de 16h. A la exploración física se encontró con desaturación y estertores crepitantes en hemitórax derecho, así como infiltrados parahiliares bilaterales en la radiografía de tórax. Se trató con cefotaxima, clindamicina y fluconazol. Por deterioro respiratorio y hemodinámico requirió de manejo en la Unidad de Terapia Intensiva Pediátrica (UTIP) con cargas de cristaloides, norepinefrina, levosimendan y ventilación mecánica por 3 días, presentando mejoría. Posteriormente presentó sepsis nosocomial y persistencia de las lesiones en cavidad bucal, por lo que se manejó con cefepima (10 días), anfotericina B (10 días) y aciclovir (7 días). Para continuar con el manejo y estudio, ingresó a medicina interna. En la valoración por Dermatología se encontró dermatosis generalizada constituida por placas eritemato-escamosas, pápulas y costras hemáticas, pruriginosas. Se indicó cetirizina y crema blanca. Se realizó tomografía computarizada (TC) (6 marzo 12) donde se encontró infiltrado retículo-nodular bilateral a nivel de bases pulmonares, hepatomegalia, múltiples adenomegalias a nivel de hueco axilar bilateral, retroperitoneales, en cadena de obturadores y en región inguinal bilateral, de hasta 2.1cm en su diámetro transverso. Es evaluado por el servicio de Oncología por incremento del tamaño y número de ganglios, hepatomegalia, fiebre vespertina-nocturna, pérdida de peso y diaforesis.
03 abril 12. En la CE de Oncología inició con prednisona a dosis de 2mg/kg/día.
24 abril 12. En la consulta de revisión persistía con adenomegalias cervicales, inguinales y axilares. La biometría hemática (BH) mostró leucocitosis (de 60,300) a expensas de neutrófilos (84%). Se continuó con prednisona.
14-18 mayo 12. En la CE de Oncología se observó una disminución importante de adenomegalias, con faringoamigdalitis aguda y lesiones bucales sugerentes de candidiasis oral. Se manejó con azitromicina y fluconazol. Con la TC de control se documentó consolidación en segmento posterior de lóbulo superior derecho, múltiples ganglios en retroperitoneo (1.4cm x 1.8cm), a nivel de ambas ilíacas (2.4cm x 2cm y 2.9cm x 1.9cm) y en región inguinofemoral (2.1cm x 1.8cm), con discreta disminución de tamaño en comparación con el estudio de marzo.
19 mayo 12. Ingresó a urgencias por epistaxis de 6 días de evolución, tos de 4 días de evolución, de inicio insidioso, no progresiva, de predominio matutino, en accesos largos, presentando expectoración verdosa, y en las últimas horas datos de dificultad respiratoria. Fiebre de 3 días de evolución hasta 40°C, con diaforesis, piloerección y escalofríos. Presentó también lesiones dolorosas en cavidad bucal de 4 días de evolución. A la exploración física presentó los siguientes signos vitales: frecuencia cardiaca 179/min, frecuencia respiratoria 26/min, tensión arterial 117/61mmHg, temperatura 39.9°C, saturación del 85%. Facies cushingoide, cavidad bucal con placas blanquecinas, induradas en carrillo derecho y encía inferior, dolorosas, no desprendibles ni sangrantes, orofaringe con descarga posterior hialina, campos pulmonares con estertores crepitantes basales derechos, adenomegalias cervicales, axilares e inguinales bilaterales, blandas, dolorosas y móviles, hígado a 4-3-3cm por debajo del reborde costal. En los estudios de laboratorio y gabinete, los resultados de la BH fueron leucocitosis de 27,600, neutrófilos 85%, bandas 8%, hemoglobina 10.5g/dl y hematocrito de 31%. Radiografía de tórax con infiltrado mixto, basal derecho y paracardiaco.
Se manejó en urgencias con ayuno, soluciones de base (1,800ml/m2SC/día, 2:1), potasio (30 mEq/m2/día), prednisona (75mg/m2SC/día), amoxicilina/sulbactam (80mg/kg/día) y fluconazol (3mg/kg/día). Se le realizaron micronebulizaciones con salbutamol, y por deterioro ventilatorio por broncoespasmo se inició ventilación mecánica no invasiva. Se progresó el esquema antibiótico a cefotaxima y dicloxacilina. Fue valorado por Oftalmología por queratoconjuntivitis de contacto, quienes iniciaron manejo con tobramicina/dexametasona tópicos.
25 mayo 12. Continuó febril. La radiografía de tórax con infiltrados algodonosos e impronta de lesiones de cavidad bucal positiva a blastoconidias. Se progresó a cefepima, amikacina y anfotericina B.
27 mayo 12. Fue valorado por Dermatología por dermatosis en tronco y región perianal, con área circunscrita de piel denudada y esfacelada de 7 días de evolución. Afectó cara a nivel de boca y mucosa bucal, con múltiples placas conformadas por úlceras con fondo blanquecino en la base de la lengua, carrillos y encías. Se inició tratamiento con agua bicarbonatada cada 6h y cicalfate tópico.
29 mayo-04 junio 12. Presentó deterioro hemodinámico y ventilatorio por broncoespasmo persistente. Requirió intubación e inicio de norepinefrina, meropenem, vancomicina y anfotericina B de complejo lipídico. Se realizó estudio histopatológico de broncoaspirado donde no se observaron hongos, bacterias ni inclusiones virales. Ingresó a la UTIP. Se retiraron aminas en 24h. La TC de tórax se observó con zonas de consolidación difusa en ambos hemitórax en “parches”, broncograma aéreo, reforzamiento heterogéneo e infiltrado alveolar difuso, a descartar hemorragia pulmonar. BH: hemoglobina 7.7g/dl, hematocrito 23.6%, leucocitos 9,600, segmentados 78%, bandas 2%, linfocitos 17%, plaquetas 231,000. Antígeno galactomanano y antígeno de Candida, negativos. Presentó salida de sangre fresca por tubo endotraqueal, y 72h después, fiebre, lesiones necrótico-hemorrágicas en labios y neumonía de focos múltiples. Se agregó trimetroprim-sulfametoxazol al manejo.
05-08 junio 12. Persistió febril, con salida de sangre fresca por tubo endotraqueal. Oncología continuó manejo exclusivo con esteroides. Nueva TC con zonas de condensación bilateral observadas en estudio previo. Múltiples adenomegalias axilares, mediastinales, en cadena ilíaca común e interna izquierda, la mayor de 2cm x 2.4cm. Conglomerados ganglionares en retroperitoneo y regiones inguinales, hepatoesplenomegalia. Continuó febril, con persistencia de adenomegalias submandibulares, axilares e inguinales. Formas invasivas en broncoaspirado: no se observaron estructuras micóticas. ELISA para VIH no reactivo. Serologías: virus Epstein Barr: IgM VCA 14.6 (negativo), IgG VCA 165 (positivo, IgG antígeno temprano 7.1, negativo), IgG EBNA 59.5 (positivo). Se extubó de manera programada.
El servicio de Cirugía de Tórax realizó endoscopia, y reportó mucosa oral muy friable, a nivel de cricofaríngeo abundantes placas blanquecinas con esfacelación de mucosa esofágica en tercio superior y medio, cambios de coloración en tercio inferior, mucosa friable sangrante. Se realizó la toma de biopsia. Se ingresó intubado a UTIP para protección de la vía aérea.
Oftalmología realizó el seguimiento del caso, observando blefaroedema, membranas verdosas en toda la superficie ocular con formación de simblefaron y córnea opaca, por lo que inició moxifloxacino y prednisona tópicos.
09-12 junio 12. Deterioro ventilatorio. Se progresó a ventilación de alta frecuencia. Se inició infusión de aminofilina, norepinefrina y milrinona. Oxemias del 68%, PVC 9 mmH2O, reserva venosa 75, Kirby 86. Sangrado por cánula orotraqueal sin mejoría en parámetros ventilatorios. Oftalmología sugiere descartar pénfigo paraneoplásico por presentar conjuntivitis folicular aguda formadora de membranas, blefaroedema con membranas abundantes y gruesas en la superficie ocular que sangraban al retirarse. BH: hemoglobina 11g/dl, hematocrito 26%, leucocitos 19,500, segmentados 89%, bandas 3%, plaquetas 398,000, TP 12.9”, TTP 36.8”, INR 1.04, fibrinógeno 495mg/dl. Creatinina 1.3mg/dl, sodio 126 mEq/l, potasio 3.8 mEq/l, calcio 6.8mg/dl, fósforo 7.9mg/dl, albúmina 1.5g/dl. Continuó con deterioro hemodinámico, sangrado fresco por nariz, boca y cánula orotraqueal, sonda urinaria con coágulos hemáticos, petequias en tórax, edema generalizado y oliguria. Radiografía de tórax con radiopacidad bilateral, en parches, y datos de hemorragia pulmonar activa. Se incrementó norepinefrina y se inició infusión de furosemide.
13 junio 12. Se inició adrenalina por persistir deterioro hemodinámico secundario a falla ventilatoria y sangrado profuso, presentó anuria y continuó hipotenso hasta progresar a paro cardiorrespiratorio. Se realizaron maniobras avanzadas de reanimación cardiopulmonar sin respuesta.
2Presentación del caso2.1Coordinadora (Dra. Martha Avilés Robles)Es importante recordar el abordaje de un paciente con adenopatías, y el momento en el que la biopsia está indicada.
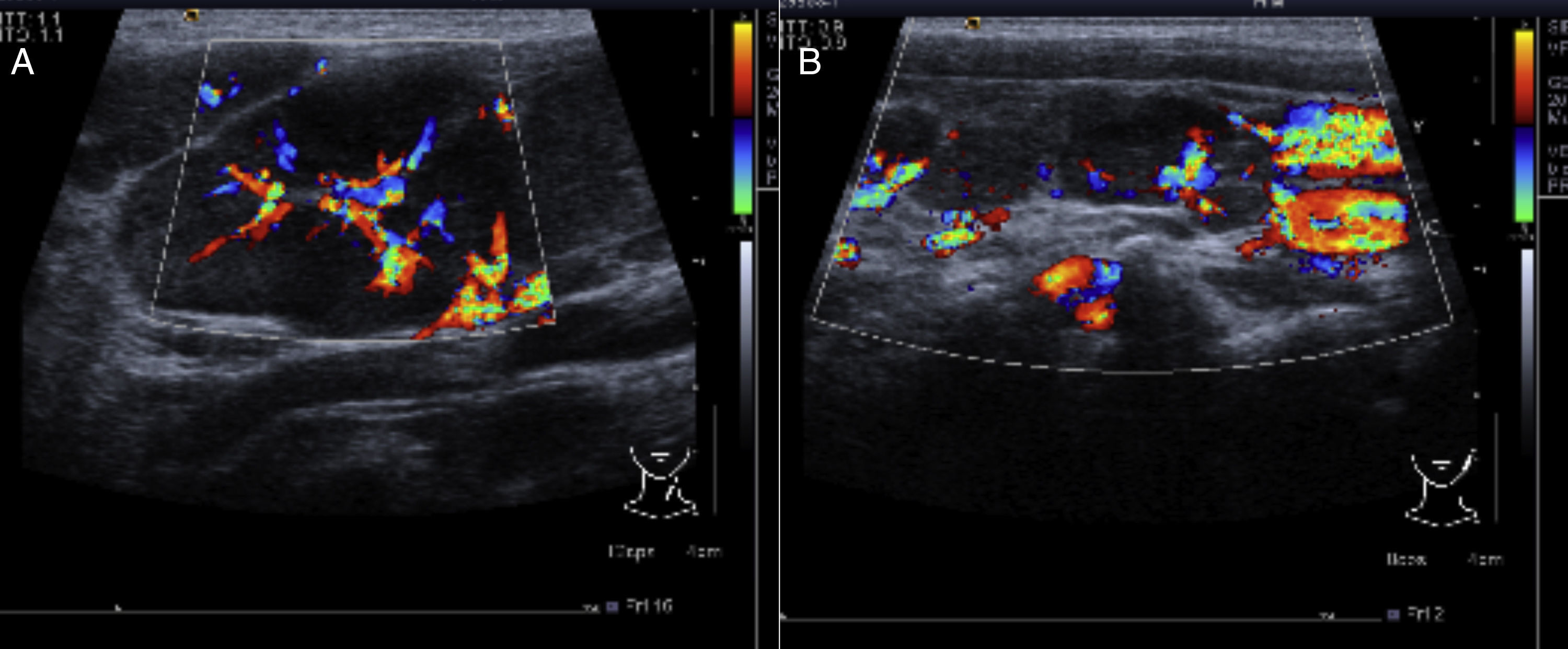
2.2Imagenología (Dra. Bertha Lilia Romero)En el ultrasonido del 23 de febrero del 2012 de la región cervical, en el lado derecho se identifican múltiples adenopatías en la cadena ganglionar. El efecto Doppler muestra un mayor flujo vascular, y el desplazamiento de los trayectos vasculares adyacentes. De lado izquierdo también se identifican adenomegalias mayores a 1cm que, al igual que el lado derecho, muestran mayor flujo vascular en su centro (Figura 1).
 Lado izquierdo. B) Lado derecho.")
Una radiografía simple de tórax del 19 de noviembre se observó de características normales. Solamente se identificó un aumento de volumen en ambas regiones cervicales en relación a crecimiento ganglionar (no se muestra figura).
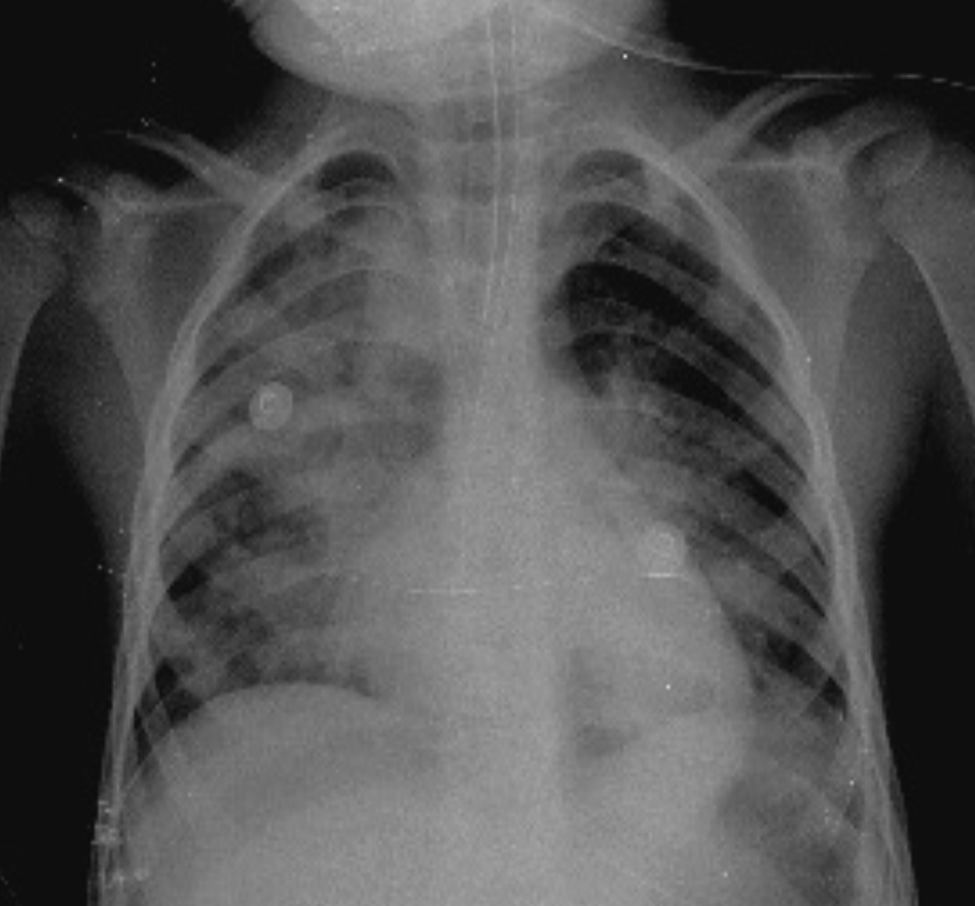
En otra radiografía se identificó radiopacidad difusa de predominio en pulmón derecho que muestra broncograma aéreo en su interior, extendiéndose desde el lóbulo apical hasta el basal del mismo pulmón, con probables zonas en región parahiliar izquierda relacionadas con el proceso neumónico. Los ángulos cardiofrénicos y costodiafragmáticos libres. Se identificó cánula en el interior de la tráquea, de adecuada posición; el resto de las estructuras no muestran alteraciones aparentes (Figura 2).

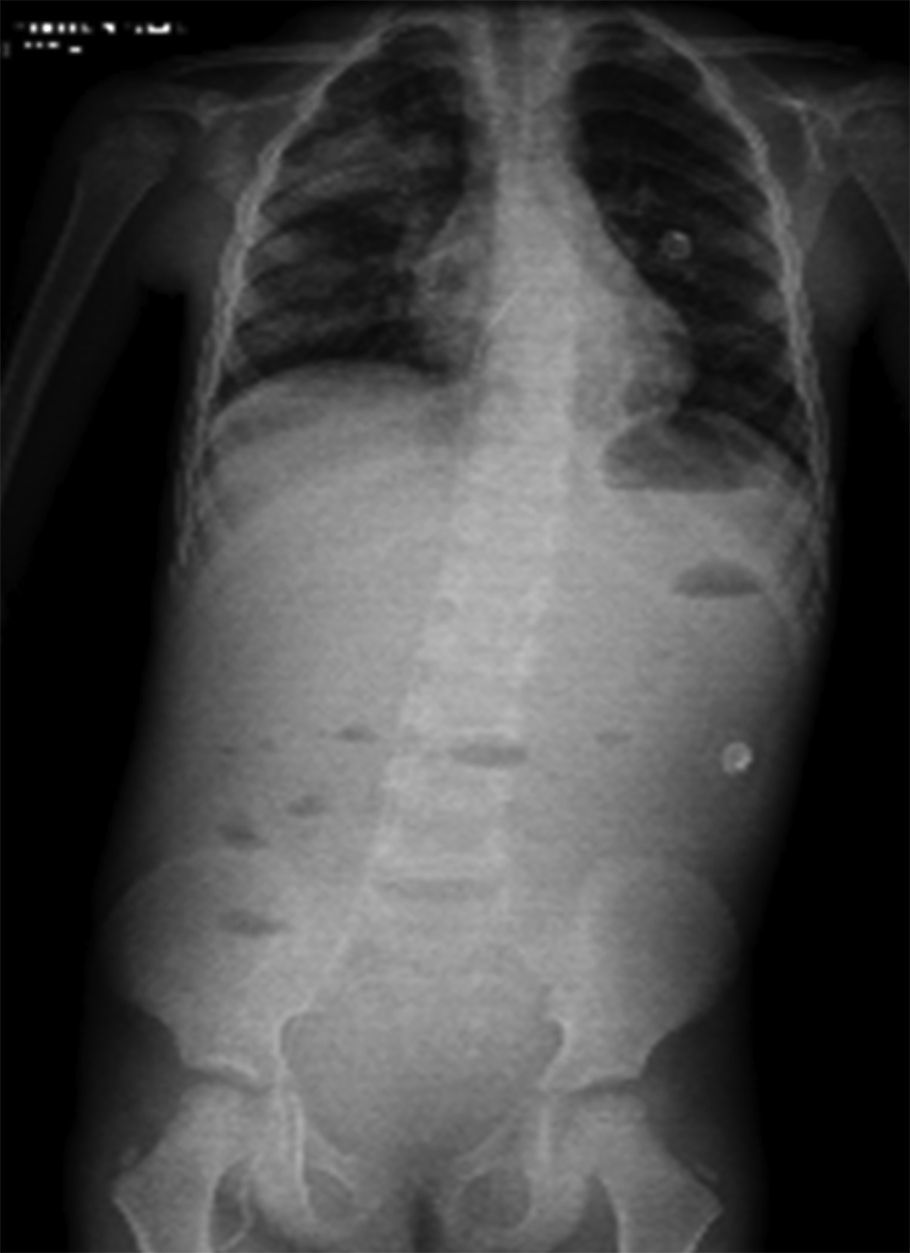
En un siguiente estudio, se tomó una proyección toracoabdominopélvica, en la que persisten las zonas de consolidación ya referidas en estudios anteriores, así como niveles hidroaéreos (Figura 3).
3Discusión3.1Urgencias (Dr. Samuel Roberts Vega)
Se trata de un preescolar masculino referido por adenomegalias, cuya biopsia de ganglio cervical fue compatible con enfermedad de Castleman. En su último ingreso consultó por fiebre, tos, dificultad respiratoria y epistaxis. Se logró integrar los siguientes diagnósticos sindromáticos:
- 1.
Síndrome de insuficiencia respiratoria. Basado en la presencia de dificultad respiratoria, estertores crepitantes basales derechos, aleteo nasal y saturación del 85%.
- 2.
Síndrome de respuesta inflamatoria sistémica. Basado en taquicardia con 179 latidos por minuto, fiebre de 40°C y leucocitosis de 27,600/mm3.
- 3.
Síndrome de inmunodeficiencia. Basado en los antecedentes patológicos que incluyen múltiples infecciones bacterianas y fúngicas, pérdida de peso y dermatosis.
- 4.
Síndrome de Cushing. Por el antecedente de administración de corticoesteroides y la facies característica.
- 5.
Síndrome mieloproliferativo. Por la presencia de adenomegalias múltiples con conglomerados ganglionares en retroperitoneo y hepatomegalia.
Luego de sustentar estos diagnósticos sindromáticos, se realizaron los siguientes diagnósticos nosológicos:
- 1.
Enfermedad de Castleman de tipo mixto o de células plasmáticas con síndrome mieloproliferativo. Inicialmente con predominio de adenomegalias cervicales y después multicéntrico, además del reporte de biopsia ganglionar.
- 2.
Neumonía adquirida en la comunidad, grave, con base en signos de insuficiencia respiratoria, desaturación, estertores crepitantes, infiltrado mixto basal y paracardiaco derecho, así como leucocitosis. Según las guías de neumonía adquirida en la comunidad en el paciente pediátrico, de la Infectious Disease Society of America, el paciente cumplió con los siguientes criterios de gravedad: hipoxemia, incremento del trabajo respiratorio, infiltrado multilobar, presencia de comorbilidad y necesidad de ventilación mecánica.
- 3.
Sepsis con base en el síndrome de respuesta inflamatoria sistémica y la presencia de una infección; en este caso a nivel pulmonar.
- 4.
Choque séptico basado en la presencia de sepsis e inestabilidad hemodinámica. Esta última sustentada por taquicardia, pulsos débiles, llenado capilar en 3 s y la necesidad de administrar aminas vasoactivas en el curso de su hospitalización.
- 5.
Hemorragia pulmonar sustentada por las imágenes radiológicas compatibles con esta entidad y el sangrado evidente a través de la cánula orotraqueal.
- 6.
Síndrome de distrés respiratorio agudo. Según los criterios de Berlín del 2011, presentó deterioro de los síntomas respiratorios, opacidades bilaterales y un índice de Kirby de 178.
- 7.
Disfunción orgánica múltiple integrada con base en el daño renal, la insuficiencia respiratoria y la inestabilidad hemodinámica; es decir, el estado de choque.
A continuación se discuten los diagnósticos y datos sobresalientes en la evolución del paciente. En primer lugar, se estuvo de acuerdo con el diagnóstico de enfermedad de Castleman, aun cuando es una entidad rara y que se presenta excepcionalmente en niños. Esta enfermedad, de acuerdo con los hallazgos histopatológicos de los ganglios afectados, fue clasificada como hialinovascular de células plasmáticas o de tipo mixto. A pesar de que en este paciente la biopsia informó un patrón hialinovascular, este tipo de enfermedad usualmente se manifiesta como unicéntrica y los pacientes cursan asintomáticos. En este caso, la evolución de la enfermedad correspondió más bien a la variedad de células plasmáticas, cuya presentación es multicéntrica y se acompaña de síntomas sistémicos tales como anemia, pérdida de peso, fiebre, hepatomegalia, esplenomegalia, hemoptisis, lesiones cutáneas, y un curso clínico más agresivo, pudiendo ser mortal sin tratamiento1–5. Ante la atípica presentación, el inicio del esteroide en la consulta externa pareció adecuado, aunque posteriormente le causa el aspecto cushingoide, pues está bien descrito como parte del tratamiento en la presentación multicéntrica. No obstante, una vez hospitalizado y evolucionando desfavorablemente, se debió considerar el uso de rituximab, que es un anticuerpo monoclonal anti-CD20 presente en las células linfoides, indicado como monoterapia o en combinación con talidomida.
Aunque la etiología de la enfermedad de Castleman es incierta, existe una asociación entre el VIH y la infección del herpes virus humano tipo 8, y este último, a su vez, con el sarcoma de Kaposi2. Las infecciones fúngicas sistémicas, infiltrados algodonosos, lesiones en cavidad oral y esófago, que si bien no pudieron ser documentadas mediante pruebas complementarias, tuvieron su valor clínico al grado de llegar a administrarle al paciente fluconazol y anfotericina B. La candidiasis oral y esofágica que manifestó pudo estar relacionada con el probable inmunocompromiso y con el uso de múltiples antibióticos en la consulta externa.
Las lesiones dérmicas descritas pueden ser compatibles con sarcoma de Kaposi, más no se tuvo una biopsia de piel que confirmara el diagnóstico, ni PCR para herpes virus humano tipo 8. Esta entidad también puede tener manifestaciones a nivel pulmonar, como disnea, sibilancias y/o hemoptisis, lo que clínicamente sería compatible con la hemorragia pulmonar que presentó el paciente.
Dentro del diagnóstico diferencial de las lesiones dérmicas, se debe incluir el pénfigo paraneoplásico. Este se presenta hasta en el 10% de los pacientes con enfermedad de Castleman. Clínicamente se caracteriza por una severa afectación oral que consiste en una estomatitis, lesiones polimorfas cutáneas que afectan preferentemente al tronco, extremidades, palmas y plantas. Puede afectar cualquier mucosa del organismo; incluso, desarrollarse una colitis pseudomembranosa grave, como la presentó este paciente. El pénfigo paraneoplásico es el único tipo clínico de pénfigo que se manifiesta en tejidos que no están tapizados con epitelio escamoso estratificado, desarrollando, en aproximadamente el 30-40% de los casos, lesiones pulmonares que pueden llevar a un desenlace fatal.
En su último internamiento, la evolución del aspecto ventilatorio fue tórpida, pues inicialmente ameritó el uso de ventilación mecánica no invasiva. En un inicio fue una buena elección puesto que el paciente la toleró durante una semana, pero posteriormente se deterioró clínicamente, presentando un estado de choque que contraindicaba la ventilación mecánica no invasiva, por lo que se decidió la intubación orotraqueal. Desarrolló síndrome de distrés respiratorio agudo. Por otro lado, aunque la clínica y la PVC podrían orientar, idealmente se debe realizar un ecocardiograma para demostrar que el edema que presentó el paciente no era cardiogénico. Probablemente desarrolló hipertensión pulmonar, dadas la hipoxemia y la necesidad de ventilación de alta frecuencia, así como la elección de milrinona en el manejo inotrópico, con la propiedad vasodilatadora pulmonar de la misma.
Hemodinámicamente presentó choque séptico por lo que requirió altas dosis de vasopresores e inotrópicos, evolucionando a disfunción orgánica múltiple con afectación del sistema circulatorio manifestado por el estado de choque, insuficiencia respiratoria evidenciada por la marcada acidosis respiratoria y el aumento de cortocircuitos pulmonares, así como insuficiencia renal aguda caracterizada por anuria, creatinina de 1.3mg/dl y una tasa de filtración glomerular de 43, tal vez combinada con hipoperfusión renal secundaria a hipovolemia por el sangrado pulmonar masivo.
Respecto al aspecto infeccioso, la cobertura inicial fue prescrita solo con base en el diagnóstico de neumonía grave y no en el escenario de un paciente séptico. La amoxicilina sulbactam tiene una cobertura muy similar a la cefotaxima, por lo que el cambio fue probablemente infructuoso. Las guías describen como terapia empírica en estos casos un beta-lactámico, usualmente cefotaxima o ceftriaxona, en adición con un aminoglucósido, esquema que debió ser el inicial. Ante la probable infección fúngica se considera que, en este paciente séptico, estaba indicada desde el inicio la cobertura con azoles, como acertadamente se realizó. Dentro del contexto del paciente con choque séptico sería lógico pensar que la hemorragia pulmonar atiende a una coagulopatía de consumo. Sin embargo, las plaquetas, los tiempos de coagulación y el fibrinógeno fueron reportados dentro de límites normales. Por ende, la causa de la hemorragia pulmonar pudo ser de las manifestaciones pulmonares del sarcoma de Kaposi, o bien de un pénfigo paraneoplásico, aunque estas teorías no pudieron confirmarse en ausencia de la biopsia de piel.
En el análisis, es difícil determinar si los signos y síntomas del paciente fueron producto de la agresiva presentación de la enfermedad de Castleman, de una inmunodeficiencia no diagnosticada, y por ende no tratada, o de ambas. No se podría aseverar que se pudo haber salvado a este paciente, pero sí se puede sugerir el inicio temprano del tratamiento para la enfermedad de Castleman multicéntrica y aumentar el índice de sospecha de inmunodeficiencias adquiridas en estos pacientes.
Diagnósticos finales:
- 1.
Enfermedad de Castleman tipo mixto o de células plasmáticas
- 2.
Probable inmunodeficiencia secundaria
- 3.
Sarcoma de Kaposi
- 4.
Pénfigo paraneoplásico
- 5.
Neumonía adquirida en la comunidad, grave
- 6.
Síndrome de distrés respiratorio agudo
- 7.
Sepsis
- 8.
Insuficiencia renal aguda
- 9.
Choque séptico
- 10.
Disfunción orgánica múltiple
Causa inmediata de la muerte: hemorragia pulmonar.
3.2Pediatría (Dra. Paloma Real Castrejón)Se identifican como adenopatías a aquellos ganglios que son>1cm. Es necesario precisar que se clasifican en localizadas y generalizadas. Las localizadas son aquellas que se encuentran en la misma región, y las generalizadas aquellas que se encuentran en dos regiones no contiguas. Es importante destacar que la etiología más frecuente de las adenopatías es la infecciosa; posteriormente, se encuentran las de tipo neoplásico, las de tipo inmunológico, manifestaciones de enfermedades endocrinológicas, y otras que se encuentran clasificadas como misceláneas. Dentro de este abordaje, es importante la historia clínica y la anamnesis: duración de las adenopatías, contacto con personas enfermas, si cuenta con un esquema de vacunación completo, si ha tenido infecciones recurrentes y síntomas constitucionales. En la exploración física se debe determinar el tamaño de las adenopatías, la localización, la infiltración de planos profundos y la consistencia. Todo esto puede orientar en el diagnóstico. Con base en los antecedentes obtenidos en el interrogatorio, se pueden solicitar algunos exámenes complementarios: biometría hemática, coombs, tiempos de coagulación, radiografía de tórax, pruebas serológicas para citomegalovirus, virus de Epstein Barr, toxoplasma, Brucella spp., factor reumatoide, antiDNA y anti-ANA. En ocasiones, la tomografía ayuda a delimitar la extensión de las adenopatías así como adenomegalias que no se pueden palpar, como las mediastinales, mesentéricas y retroperitoneales.
Se debe realizar una biopsia de aquellas adenopatías>3cm, y de aquellas que se presentan en un paciente con síntomas constitucionales, así como las que presentan consistencia dura, que estén infiltrando tejidos profundos y que tengan una localización supraclavicular.
Es importante recordar que estos estudios no se solicitan todos al mismo tiempo, y que el interrogatorio clínico y la exploración física son los que van a orientar sobre qué estudios se deben solicitar de primera instancia.
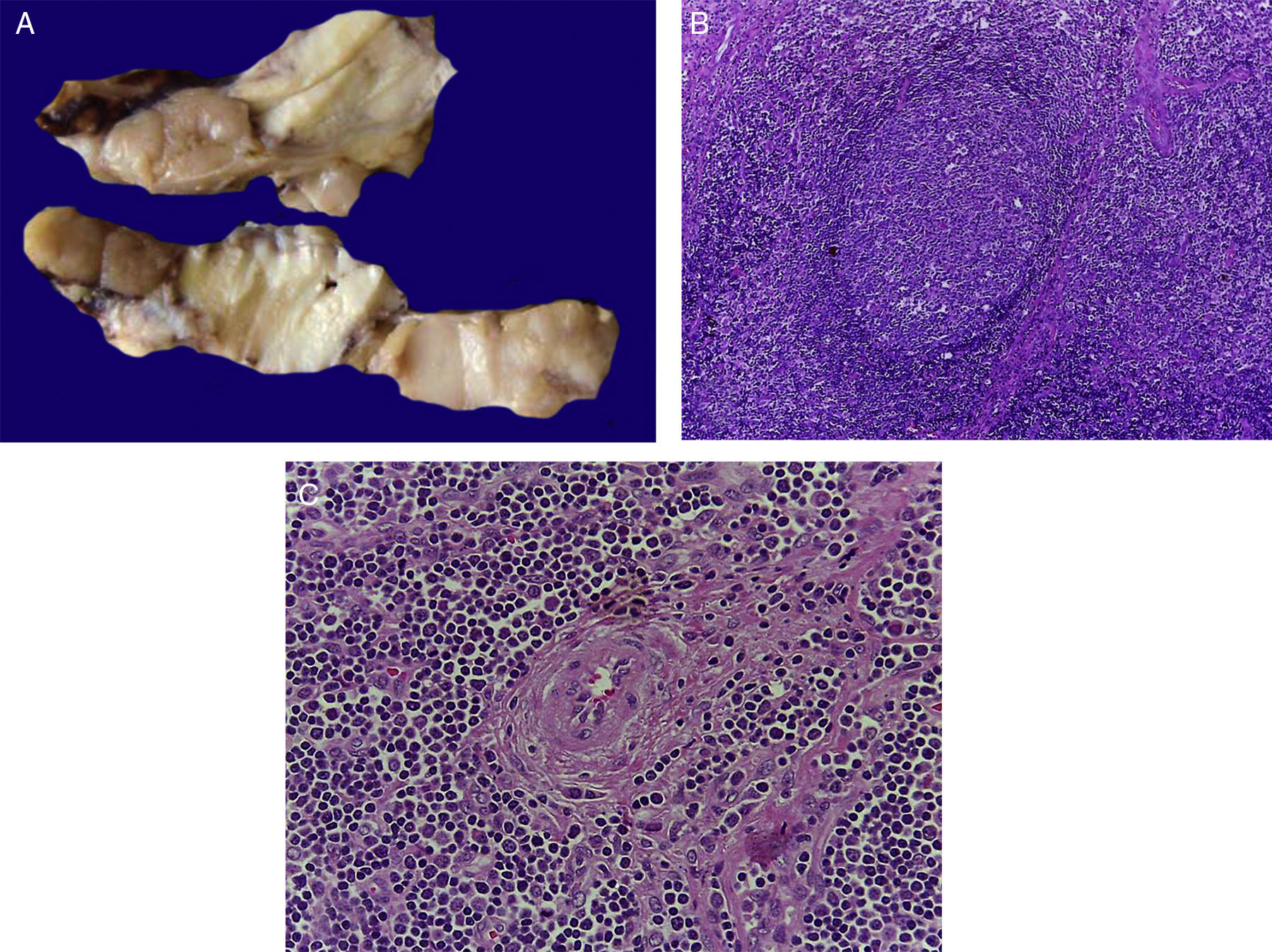
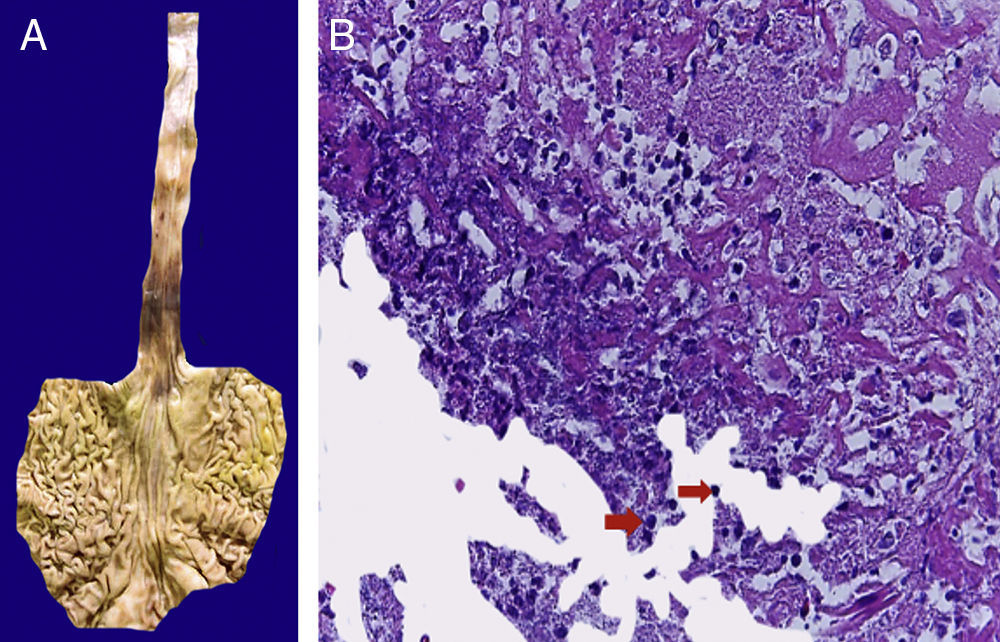
4Patología (Dr. Mario Perezpeña-Diazconti)El primer contacto que tuvimos con el paciente fue el 30 de noviembre del 2011 en el que recibimos un ganglio linfático aumentado de tamaño que midió 4cm de eje mayor (Figura 4A). Histológicamente, existe expansión de la zona paracortical con grandes folículos linfoides de tamaño variable, aumentados en número, con disposición concéntrica de los linfocitos que formaban lo que se describe como tela de cebolla, con centros germinales activos (Figura 4B). Los vasos muestran hiperplasia de la pared y proliferación concéntrica del tejido fibroconectivo (Figura 4C). Se realizaron estudios de inmunohistoquímica (CD20) en los que resaltó la población de linfocitos B en el centro germinal (como es normal en los ganglios linfáticos), y de linfocitos T alrededor de estos centros germinales. No se observaron datos de neoplasia maligna. Se diagnosticó como enfermedad de Castleman de variedad hialino vascular.
. Hiperplasia folicular con la disposición concéntrica de linfocitos en la periferia (B). Los vasos muestran hiperplasia de la pared (C).")
La enfermedad de Castleman, o hiperplasia de ganglios linfáticos gigantes, se representa en esta foto macroscópica, con el notorio crecimiento del conglomerado de ganglios linfáticos. En el estudio post mórtem, midieron 3 a 4cm de eje mayor cada uno (A). Hiperplasia folicular con la disposición concéntrica de linfocitos en la periferia (B). Los vasos muestran hiperplasia de la pared (C).
Los resultados de la Inmunohistoquímica fueron los siguientes:
Ganglio linfático cervical
- •
CD10: positiva en centros germinales
- •
CD20: positiva en la corteza
- •
CD3: positiva en la paracorteza
- •
LMP1: negativa
- •
CD30: negativa
- •
BCL-6: positiva en los centros germinales
Posteriormente, el 28 de diciembre del 2011 se recibió otro ganglio linfático. El aspecto histológico fue similar al recibido anteriormente (Figura 4). El diagnóstico se confirmó como enfermedad de Castleman variedad hialino vascular. El 23 de febrero del 2012 se recibió una citología de vesículas en la cavidad oral. Se informó que había células inflamatorias con mala conservación, cambios por desecación y sin inclusiones virales. Nuevamente, el 09 de marzo del 2012 se recibió otro ganglio linfático, en el que, a diferencia de los ganglios previos, hubo pérdida de la morfología ya que no fue posible determinar la zona folicular ni la paracorteza, y se observó un patrón en cielo estrellado, con macrófagos con cuerpos teñibles. Se hicieron reacciones de inmunohistoquímica con CD20 positivo en el centro germinal, las áreas de linfocitos T positivas para CD3 y, en este caso, se hizo también un CD138 para demostrar la presencia de células plasmáticas, presentes en la variedad de enfermedad de Castleman, que fueron muy escasas. Se hizo el diagnóstico de hiperplasia paracortical difusa, negativa para células neoplásicas. Posteriormente se recibió un broncoaspirado, con escasas células inflamatorias. No se observaron células de epitelio respiratorio ni Aspergillus sp., pneumocistis o bacterias.
El 08 de junio del 2012 se recibió una biopsia de esófago con áreas ulceradas de la mucosa con infiltrado inflamatorio, fibrina y detritus celular. No se observaron microorganismos. El diagnóstico fue de una esofagitis aguda ulcerada.
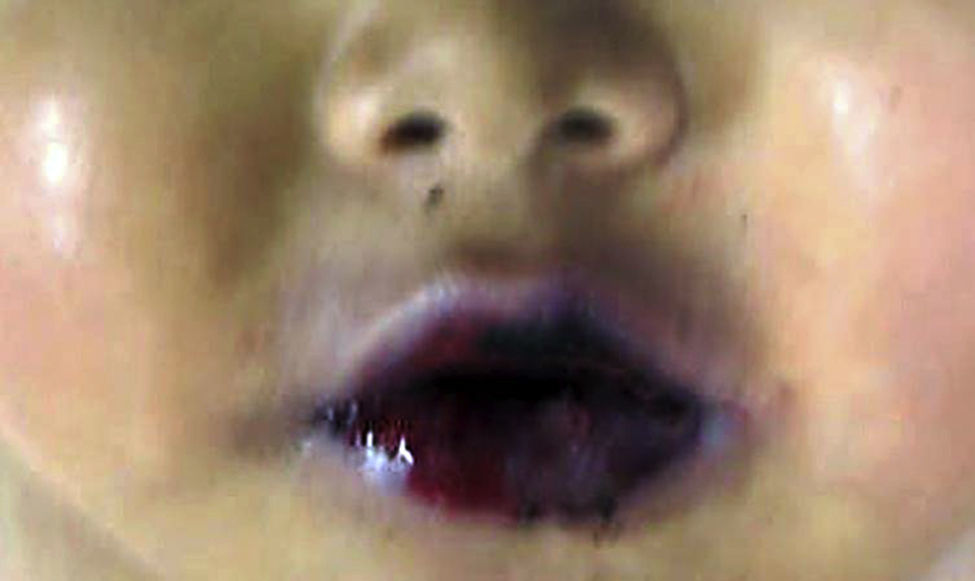
4.1Estudio post mórtemEl paciente presentó desnutrición, lesiones ulceradas y ampollas en los labios así como en la cavidad oral (Figura 5). En el cuerpo no se observó ninguna lesión ampollosa. Presentó manchas de color café mal delimitadas, de tamaño variable y que ocupaban prácticamente toda la cara anterior del tórax y del abdomen. A la apertura de cavidades se encontró líquido serohemático en cavidades pleurales, 50ml de cada lado. En la cavidad abdominal se encontraron 40ml de líquido serohemático. Las cadenas ganglionares estaban muy aumentadas de tamaño. Cada ganglio medía de 2 a 4cm, y afectaban todas las cadenas ganglionares; algunos de estos mostraban necrosis extensa. Histológicamente se observó necrosis completa del ganglio linfático o pérdida de la morfología sin necrosis. En los ganglios se hicieron tinciones de histoquímica para buscar hongos o bacterias, pero fueron negativas.

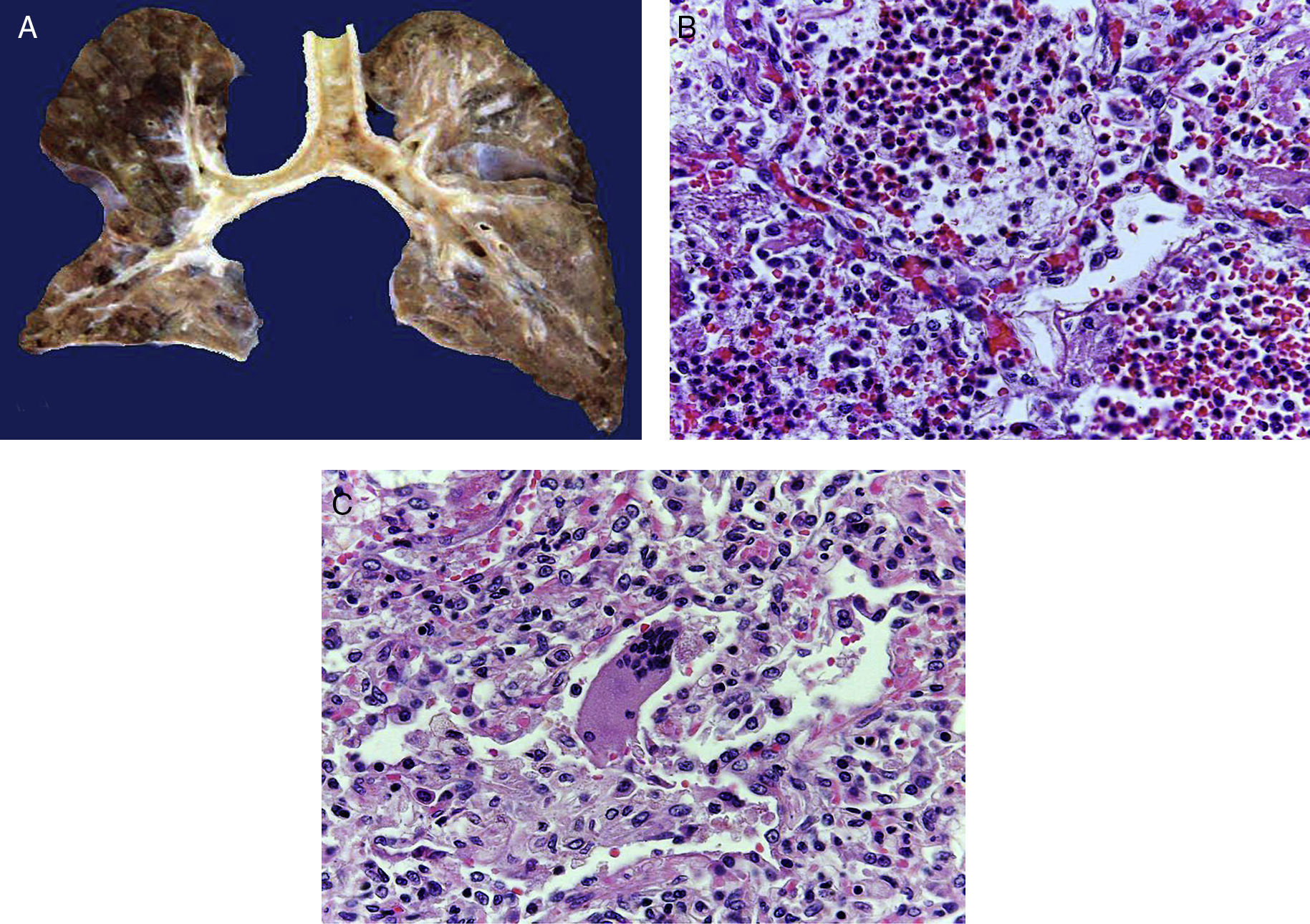
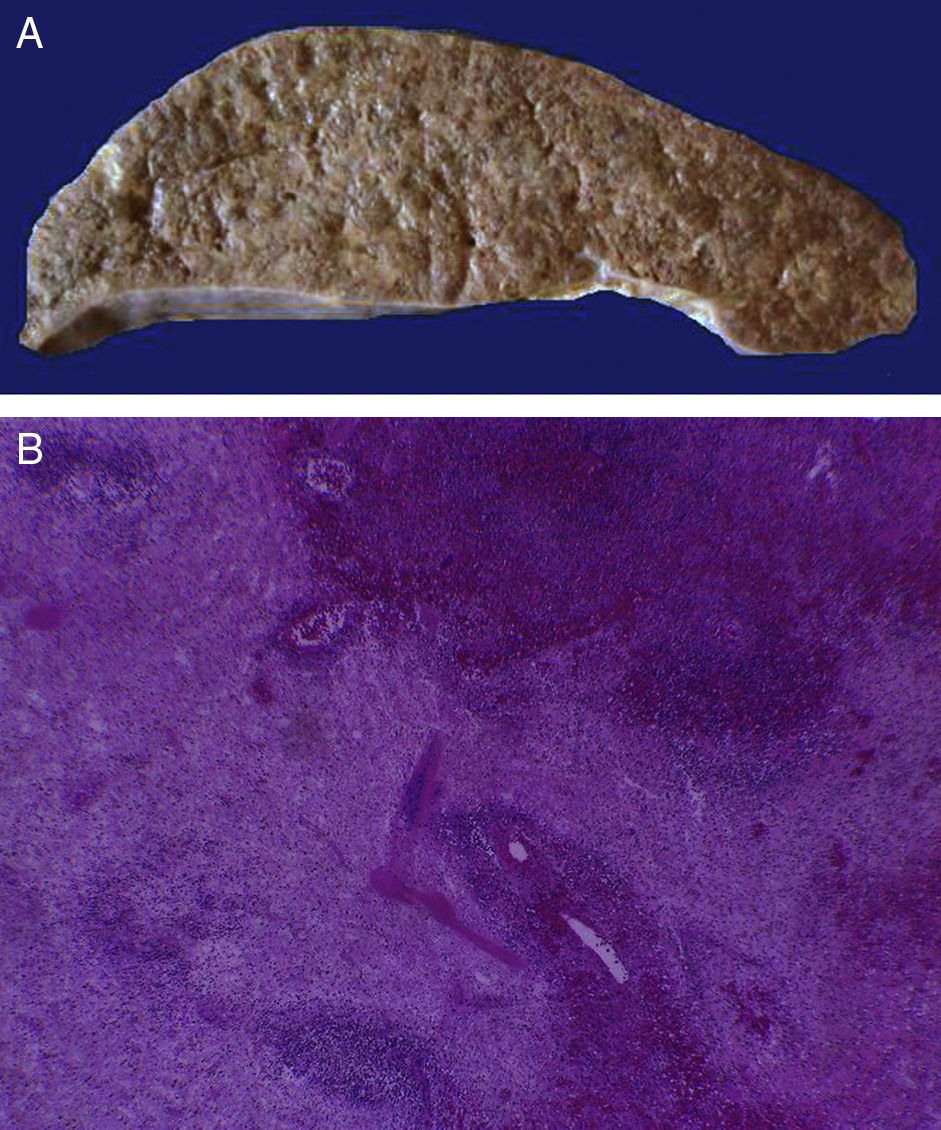
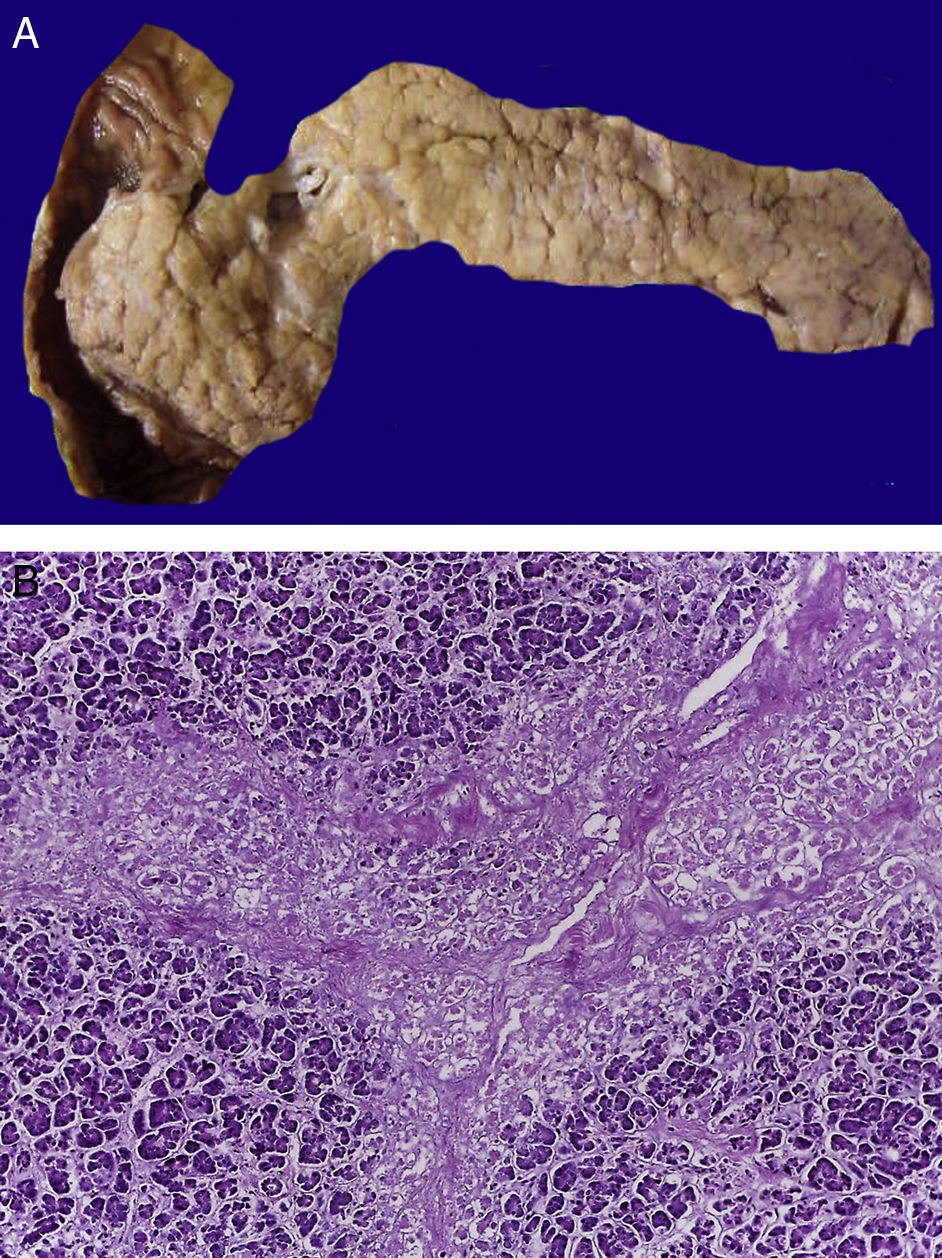
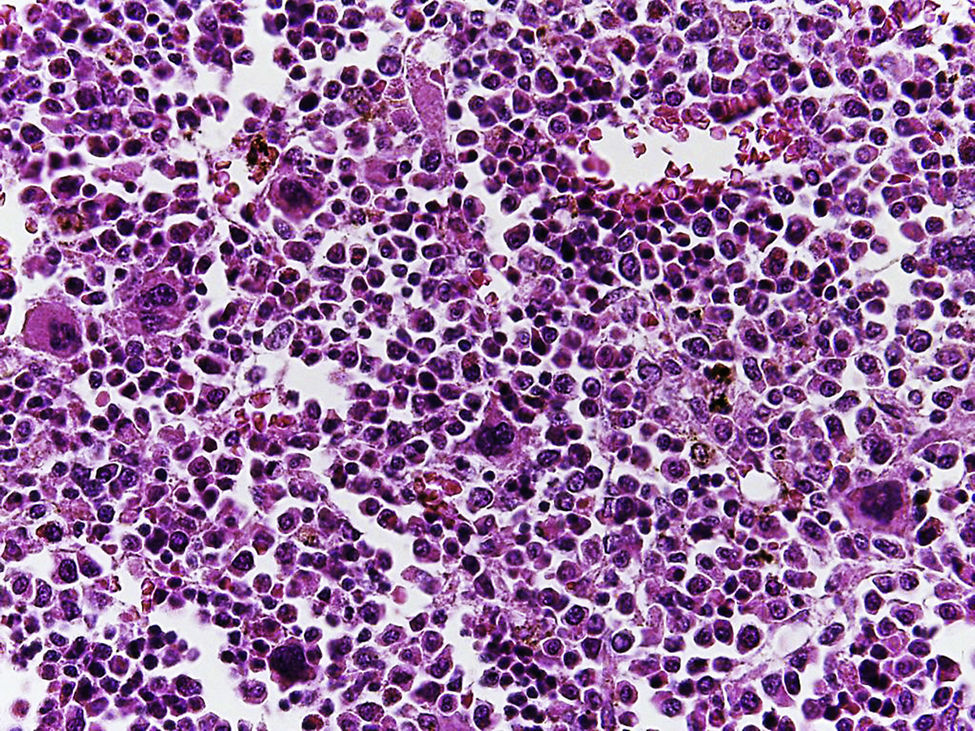
En la tráquea, se encontró gran cantidad de detritus celular y un coágulo sanguíneo que obstruía completamente la luz (Figura 6A). Los pulmones pesaron 250g, con aumento de tamaño y peso. Histológicamente, la mucosa de los bronquios principales mostró el epitelio escamoso ulcerado, con gran cantidad de infiltrado inflamatorio, detritus celular y necrosis (Figura 6B). En el parénquima pulmonar se observó hemorragia y formación de microabscesos, formación de membranas hialinas debido a daño multifactorial y focalmente células gigantes multinucleadas que sugieren una infección viral (Figura 6C). El bazo se encontró aumentado de tamaño y peso (Figura 7A), con aspecto nodular y extensas zonas de infartos isquémicos relacionado al estado de choque o sepsis (Figura 7B). El hígado se encontró aumentado de tamaño y de peso, histológicamente con congestión y sin otra alteración. El páncreas estaba aumentado de tamaño y de peso (Figura 8A), histológicamente encontramos zonas de necrosis del páncreas acinar (Figura 8B). La mucosa del esófago y el estómago muestran úlceras de tamaño variable. La mayor afecta el epitelio en el tercio medio y el tercio inferior del esófago, incluyendo la unión con el estómago (Figura 9A). Histológicamente, la mucosa ha desaparecido y ha sido reemplazada por fibrina, detritus celular y leucocitos polimorfonucleares e infiltrado inflamatorio por debajo de la mucosa, y la presencia de algunas células multinucleadas que sugieren que hay alguna infección de tipo viral. Este daño se extendía hasta la unión esófago-gástrica y mucosa gástrica. En la laringe, la mucosa está ulcerada con infiltrado inflamatorio abundante y células que sugieren proceso viral. Además, se encontró la presencia de pequeñas estructuras esféricas que corresponden a levaduras Candida sp. (Figura 9B). De acuerdo con la historia clínica había disminución en el número de plaquetas. En la médula ósea había aumento en el número de megacariocitos; sin embargo, estos no presentaban alteraciones morfológicas y no se observó mielofibrosis (Figura 10).
. Histológicamente los espacios alveolares están ocupados por leucocitos polimorfonucleares, incluyendo abundantes eosinófilos y macrófagos (B). En algunas zonas hay células gigantes multinucleadas que sugieren infección viral (C).")
La luz de la tráquea está ocupada por fibrina, detritus celular y material hemático. Al corte, los bronquios principales muestran ulceración de la mucosa, el parénquima es de color café rojizo, de aspecto sólido en zonas hemorrágicas (A). Histológicamente los espacios alveolares están ocupados por leucocitos polimorfonucleares, incluyendo abundantes eosinófilos y macrófagos (B). En algunas zonas hay células gigantes multinucleadas que sugieren infección viral (C).
. Histológicamente se observan extensas zonas de necrosis asociadas al estado de choque (B).")
 y del componente acinar (B).")
. Histológicamente, la ulcera está cubierta por fibrina, detritus celular y estructuras esféricas que corresponden a levaduras de Candida sp. (flechas), y células gigantes multinucleadas, como las observadas en el pulmón (B).")
Estómago y esófago. La mucosa de ambos muestra extensa ulceración que afectan el tercio medio e inferior del esófago y la unión esófago-gástrica (A). Histológicamente, la ulcera está cubierta por fibrina, detritus celular y estructuras esféricas que corresponden a levaduras de Candida sp. (flechas), y células gigantes multinucleadas, como las observadas en el pulmón (B).

El diagnóstico principal es enfermedad de Castleman hialino vascular tratada con esteroides. La necrosis que se observó en los ganglios en el estudio post mórtem fue debida al tratamiento.
4.2Diagnósticos anatómicos finalesEnfermedad de Castleman hialino vascular tratada con esteroides, con linfadenitis aguda necrosante no específica que afectó ganglios linfáticos cervicales, mediastinales, axilares, retroperitoneales, abdominales e inguinales6–10.
4.3Alteraciones concomitantes- •
Bronconeumonía aguda mixta con cambios citopáticos, que sugieren infección viral e infección bacteriana
- •
Daño alveolar difuso multifactorial por sepsis y estado de choque
- •
Hemorragia pulmonar extensa
- •
Neumotórax bilateral (50ml derecho e izquierdo)
- •
Mucositis con glositis y laringitis aguda ulcerada, con cambios citológicos que sugieren infección viral
- •
Esofagitis aguda ulcerada que afectó tercio medio y distal, con cambios citológicos que sugieren infección viral
- •
Hepatomegalia congestiva (PO 1100g/PE 588g kayser)
- •
Esplenomegalia (PO 120g/PE 58g kayser)
- •
Nefromegalia (PO 180g/PE 110g kayser)
- •
Pulmones (PO 500g/PE 275g kayser)
- •
Suprarrenales (PO 10g/PE 7.5g kayser)
- •
Liquido en cavidad peritoneal (40ml serohemático)
- •
Infartos recientes en bazo
- •
Pancreatitis aguda
- •
Hemocultivo, cultivo de pulmón derecho e izquierdo, hígado e intestino sin desarrollo de bacterias
- •
LCR: escaso desarrollo de Enterococcus faecium
- •
Bazo: escaso desarrollo de Enterococcus faecium y Enterococcus sp
- •
Colon: escaso desarrollo de Enterococcus faecium, Klebsiella pneumoniae y dos morfotipos de E. Coli
La enfermedad de Castleman presenta dos variedades histológicas: la hialino vascular y la de células plasmáticas (multicéntrica). Así mismo, pueden tener diferentes formas de presentación clínica: 1) única (puede ser un solo ganglio afectado, y al realizar una extirpación el paciente está prácticamente curado); 2) multicéntrica (es la forma grave que casi siempre lleva a la muerte, que es la que presentó este paciente).
La enfermedad de Castleman multicéntrica o linfadenopatía plasmática idiopática se divide en dos grandes grupos con características clínicas diferentes. Como en este caso, los pacientes presentan derrame pleural, ascitis, linfadenopatía sistémica, trombocitopenia y enfermedades autoinmunes cuando tienen el elemento de hiperinmunoglobulinemia policlonal. Los pacientes sin hiperinmunoglobulinemia policlonal presentan trombocitopenia, anasarca/fiebre, fibrosis reticulínica en la médula ósea, organomegalia, EC-HV y trombocitopenia menor a 100,000 plaquetas.
De acuerdo con lo anteriormente expuesto, el paciente que se presentó fue portador de enfermedad de Castleman multifocal de variedad hiperinmunoglobulinemia policlonal.
5Comentarios finales5.1Coordinadora (Dra. Martha Avilés Robles)Cuando la enfermedad de Castleman es de tipo localizada, el tratamiento quirúrgico resuelve la enfermedad. Sin embargo, cuando la enfermedad es multicéntrica, la respuesta generalmente es inadecuada y se requieren diferentes maniobras o estrategias de tratamiento.
5.2Oncología (Dra. Elisa Dorantes Acosta)Lo primero que se debe hacer es sospechar el diagnóstico. Hay que pensar en una enfermedad linfoproliferativa muy rara; de hecho, en los textos no se conoce exactamente la incidencia. Se conoce que es más frecuente en la edad adulta, pero la incidencia en la edad pediátrica no está descrita adecuadamente.
Retomando los comentarios anteriores, se debe considerar si se tiene un ganglio de dimensiones incrementadas de tamaño, si ya se dio una terapia estándar con aintiinflamatorios no esteroideos (AINES) y no resuelve, o si está en alguna zona que se considera terreno patológico; entonces, se debe realizar una biopsia. Después se tiene que definir qué tipo de biopsia. Sobre todo en pediatría, la biopsia debe ser excisional. Esto es muy importante para que Patología pueda observar toda la arquitectura del ganglio. En los reportes que hay de casos de enfermedad de Castleman, existe el problema que se hacen biopsias por aspiración de aguja fina y terminan haciendo la escisión del ganglio. De hecho, si la enfermedad hubiera sido de tipo localizada, tan solo con la extirpación quirúrgica del ganglio se hubiera curado el niño. Sin embargo, en este caso en el que la enfermedad era multicéntrica o sistémica, se debieron hacer varias consideraciones:
- 1.
El diagnóstico de enfermedad de Castleman se realiza por patología. Al sospechar de un ganglio anormal, el patólogo puede dar el diagnóstico de la enfermedad de Castleman porque va a diferenciar entre un proceso maligno y uno como este, que aunque no fue cáncer pudo asociarse con varias cosas. Esta enfermedad no es aislada, por lo que se debe pensar en inmunodeficiencias, en asociaciones con algunos tipos de virus, etcétera. Se deben realizar serologías virales cuando se tiene el reporte de enfermedad de Castleman, para poder efectuar un diagnóstico preciso.
- 2.
Si es sistémico, lo primero que se debe ver es si alguno de los ganglios está ocasionando problemas que pongan en riesgo la vida del paciente. Hay descripciones de pacientes con masas mediastinales extensas que ocasionan problemas respiratorios o de retorno circulatorio en los que se debe iniciar la terapia esteroidea de emergencia8. En algunos otros reportes o en algunos otros centros se considera incluso la radioterapia localizada en aquellos ganglios que están poniendo la vida del paciente en riesgo, aunque sea enfermedad sistémica.
Una vez que se ha evaluado si hay riesgo por algún ganglio que esté comprometiendo la vida del niño, independientemente de la enfermedad de Castleman, a continuación se debe considerar qué se va a intentar como terapia de primer abordaje. No existen guías de manejo para esta enfermedad, y la experiencia con que se cuenta es de dos casos aislados: se ha probado el uso de prednisona por 2 meses y después la reducción con anticuerpos monoclonales, con regulares resultados. Por otro lado, también se ha intentado la terapia con alcaloides de la vinca. Así mismo, se recomienda retirar quirúrgicamente el ganglio que más afecta y radiar. La terapia es diversa ya que no hay un acuerdo internacional. Pero se podría resolver siguiendo, como pediatra, el abordaje de un ganglio patológico.
Se consideró que este paciente tenía una enfermedad sistémica de variedad hialino vascular, pues se descartaron otros factores que pudieran asociarse. Sin embargo, no se descartó el conjunto sindromático POEMS: polirradiculoneuropatía, que no se incluyó en el resumen clínico pero se pudo preguntar de manera intencionada; organomegalia, que sí tenía; endocrinopatía, que no se abordó; monoclonalidad, que no tenía por lo menos de un linfoma; y alteraciones en la piel (skin), que sí tenía. El paciente contaba con algunos datos probablemente relacionados con este conjunto sindromático de afecciones múltiples que faltó descartar. Dada la rareza del diagnóstico, se debió considerar como diagnóstico diferencial y descartar estas otras alteraciones que no se mencionaron en el resumen clínico están y que podrían haberse corregido. No se abordó con profundidad la posibilidad de que el niño tuviera alguna inmunodeficiencia primaria, aunque una secundaria (VIH) no tenía. Sin embargo, era necesario este abordaje para realizar el conjunto diagnóstico, y probablemente ofrecer alguna otra terapéutica.
5.3Coordinadora (Dra. Martha Avilés Robles)Durante el curso de su enfermedad este paciente desarrolló un pénfigo paraneoplásico que se sospechó hasta una etapa tardía de la enfermedad. Es importante recordar sus formas de presentación para sospechar oportunamente el diagnóstico.
5.4Dermatología (Dr. Carlos Mena Cedillos)El pénfigo paraneoplásico es una enfermedad rara de los ganglios linfáticos y tejidos relacionados. Aunque no es cáncer, es considerado un desorden linfoproliferativo, e incluso algunas formas multicéntricas pueden comportarse de manera agresiva y requerir tratamiento con quimioterapia y/o radioterapia11,12.
La participación de Dermatología en realidad no viene a ser tan directa en cuanto a los síntomas generales. Se observó que, al momento del ingreso, el niño tenía manifestaciones muy inespecíficas. Incluso era una dermatosis eritematoescamosa-papular, que en realidad no es una descripción diagnóstica per se. Fue abordado como una dermatosis reactiva con diversas posibles lesiones, como se reporta en la literatura: ampollas flácidas o tensas, con o sin erosiones (que en el paciente, en un segundo ingreso, se observaron esfaceladas), con lesiones de tipo eritema multiforme y de tipo liquen plano. Este polimorfismo puede estar presente en el mismo paciente, en un mismo momento (como en este caso), como también puede ser a lo largo de su evolución: en el primer ingreso, el paciente presentó lesiones eritematoescamosas, y en el segundo ingreso ya eran más evidentes las lesiones ampollares, agregándose estomatitis.
En los niños es más frecuente encontrar una dermatitis liquenoide en tronco y extremidades, a diferencia de lo que se menciona en el adulto, que fue lo que se observó en el primer ingreso de este niño. Incluso es común encontrar el compromiso palmoplantar y afectación ungueal.
En cuanto al pénfigo paraneoplásico, es quizás la parte más relevante de la expresión dermatológica. Desafortunadamente, en este caso no se pudo documentar en vida. Es un hallazgo constante, e incluso el primer signo de la enfermedad, el compromiso de la mucosa oral. Las erosiones y ulceraciones dolorosas que afectan la mucosa y se extienden hasta el bermellón de los labios son características, de difícil manejo y refractarias al tratamiento. También hay compromiso de conjuntivas, mucosa esofágica y anogenital.
Esto son los criterios de Anhalt de pénfigo paraneoplásico12:
- •
Clínicos: erosiones dolorosas de las mucosas, con lesiones cutáneas polimorfas asociadas con una neoplasia
- •
Hallazgos histológicos: acantólisis intraepidérmica, queratinocitos necróticos, dermatitis de interfase vacuolar
- •
Inmunofluorescencia directa: depósitos de complemento e IgG en espacios intercelulares del epitelio y en forma variable a lo largo de la membrana basal
- •
Inmunofluorescencia indirecta: presencia de anticuerpos circulantes que reconocen espacios intercelulares del epitelio estratificado y transicional
- •
Inmunoprecipitación de anticuerpos séricos frente a antígenos de alto peso molecular: desmoplaquina 1 (250kD), antígeno de penfigoide ampolloso (230kD), desmoplaquina 2 y envoplaquina (210kD), periplaquina (190kD) y un antígeno no identificado de 170kD
Por otro lado, los criterios diagnósticos de Camisa y Helm incluyen tres criterios mayores —erupción mucocutánea polimorfa, neoplasia interna asociada, test de inmunoprecipitación característico— y tres criterios menores: acantólisis en la histopatología, inmunofluorescencia directa perilesional con patrón intracelular y en la zona de la membrana basal, inmunofluorescencia indirecta en vejiga de rata positiva. Se requiere cumplir los tres criterios mayores o dos mayores y dos menores13.
En la publicación de Cervini y colaboradores se hace referencia a una niña de 10 años con enfermedad de Castleman y el pénfigo paraneoplásico y a otro niño de 12 años con pénfigo paraneoplásico asociado con linfoma de Hodgkin13. Es muy relevante que cuando el médico se enfrente a la enfermedad de Castleman con pénfigo paraneoplásico se realicen los diagnósticos diferenciales, sobre todo con patologías que cursen con estomatitis refractarias, en este caso ante la comprobación de la presencia de Candida en mucosa. Esto pudo retrasar el diagnóstico de pénfigo paraneoplásico por la expresión que tenía a nivel de mucosas, pero también se debe distinguir de eritema multiforme, de síndrome de Steven Johnson, necrólisis epidérmica tóxica, pénfigo vulgar sin asociación con neoplasias, penfigoide ampolloso, liquen plano, infecciones herpéticas, entre otras.
Este caso se pudo haber demostrado mediante pruebas inmunológicas.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.