





El hipotiroidismo congénito (HC) es una causa prevenible de retraso mental, por lo que es de suma importancia que el diagnóstico y tratamiento oportunos sean realizados por el médico de primer contacto. El tamizaje para HC se debe realizar entre el segundo y quinto días de vida, con sangre capilar mediante la punción del talón. Debe confirmarse mediante el perfil tiroideo en sangre venosa.
La etiología más frecuente es la disgenesia tiroidea, la cual se identifica con gamagrafía tiroidea antes de iniciar el tratamiento. El tratamiento es con levotiroxina (10-15μg/kg/día) y no debe ser retrasado ni suspendido durante los tres primeros años de vida debido al efecto deletéreo en el neurodesarrollo en el caso de deficiencia de hormonas tiroideas durante esta etapa. Los recién nacidos prematuros, enfermos o con síndrome de Down requieren una valoración especial. En este artículo se describen los algoritmos diagnósticos y terapéuticos del HC.
Congenital hypothyroidism (CH) is a cause of preventable mental retardation; therefore, timely diagnosis and treatment by the primary care physician is very important. CH screening must be performed between the second and fifth days of life with capillary blood done with a heel prick and must be confirmed by measurement of thyroid hormones in venous blood.
The most common cause of CH is thyroid dysgenesis, which may be identified by a thyroid scan carried out before initiating treatment. Treatment should be with levothyroxine (10-15μg/kg/day) and should not be delayed or suspended during the first 3 years of life due to the deleterious effect on neurodevelopment in case of low thyroid hormones during this time. Preterm or sick infants or those with Down syndrome require special consideration. This article provides diagnostic and therapeutic algorithms for CH.
El hipotiroidismo congénito (HC) es la deficiencia de hormonas tiroideas presente al nacimiento. El HC es una causa de retraso mental prevenible. Por lo general no presenta signos o síntomas floridos en el momento del nacimiento, pero el pronóstico neurológico depende del inicio oportuno y correcto del tratamiento. De ahí la importancia del diagnóstico temprano mediante el tamiz neonatal.
2EpidemiologíaLa frecuencia de HC varía de acuerdo con diversos factores: 1) el área geográfica; 2) la frecuencia de deficiencia de yodo en la población; 3) el periodo de estudio; 4) la metodología utilizada para el tamizaje; y 5) las concentraciones de hormonas seleccionadas como puntos de corte para el diagnóstico. En el mundo, la prevalencia se ha estimado entre 1:800-1:10,000; en México se estima en 1:2,4001. El HC es más frecuente en asiáticos, hispanos e indígenas americanos en comparación con la población blanca o afroamericana; presenta un predominio en mujeres con una relación de 2:1 a 3:12.
3EtiologíaLa principal causa de HC es la deficiencia de yodo3. En las regiones con suficiencia de yodo, la mayoría de los casos de HC son esporádicos. De estos, a su vez, la mayoría son por disgenesia tiroidea, es decir, alteraciones en la morfogénesis de la tiroides; en solo el 2% de los casos hay agregación familiar4. En México se ha reportado que el 57% de los casos detectados por tamiz se deben a ectopia tiroidea, el 36% a agenesia tiroidea y el 7% a dishormogénesis5. En la tabla 1 se muestran las causas de hipotiroidismo congénito permanente6.
Causas de hipotiroidismo congénito permanente
| I. Primario (¿ 1:2,400) | |
|---|---|
| 1. No sindrómico | 2. Sindrómico |
| a. Disgenesia o disembriogénesis (75-90%) | a. Mutación de NKX2.1/TTF1 (HC, corea y/o ataxia, neumopatía) |
| Ectopia (40%) | b. Mutación de TTF2 (síndrome de Bamforth-Lazarus: disgenesia tiroidea, atresia de coanas, paladar hendido, pelo puntiagudo) |
| Agenesia/atirosis (30-40%) | c. Mutación de PAX8 (asociado con malformaciones urinarias) |
| Hipoplasia (5%) | d. Mutación de NKX2.5 |
| Hemiagenesia (< 5%) | e. Mutación de proteína Gsα (osteodistrofia hereditaria de Albright) |
| b. Dishormogénesis (¿ 20%) | f. Síndrome de Pendred (bocio e hipoacusia) |
| Mutación del cotransportador de sodio-yodo | g. Displasia ectodérmica (HC, hipohidrosis, disciensiaciliar) |
| Deficiencia de tiroglobulina | h. Hipotiroidismo-dismorfismo, polidactiliapostaxial, déficit intelctual |
| Defectos de peroxidación (DUOX2 y DUOXA2) | i. Síndrome de Kocher-Deber-Semilange (psedohipertrofia muscular e hipotiroidismo) |
| Deficiencia de pendrina | j. Obesidad-colitis-hipotiroidismo-hipertrofia cardiaca-retraso en el desarrollo |
| Defecto en desyodasas (DEHALI y SECISBP2) | |
| c. Exposición materna a radioyodo | |
| d. Mutación del receptor de TSH (<5%)15 | |
| e. Mutación inactivadora de proteína Gs | |
| II. Central (1:50,000-1:100,000) | III. Periférico |
| 1. Mutación de subunidad beta de TSH | 1. Resistencia a hormonas tiroideas |
| 2. Deficiencia de TRH (deficiencia aislada/interrupción del tallo hipofisiario/lesión hipotalámica) | 2. Mutación de MCT8 (síndrome de Allan-Herndon-Dudley) |
| 3. Resistencia a TRH | |
| 4. Defecto en factores de trascripción de desarrollo hipofisiario | |
| 5. Mutación de IGSF116 |
DUOX2: dual oxidase 2; DUOXA2: dual oxidase 2 maturation factor; DEHALI: iodotirosine deshalogenase; SECISBP2: sec insertion sequence binding protein 2; TSH: hormona estimulante de la tiroides; TRH: hormona liberadora de tirotropina; IGSF1: immunoglobulin superfamily 1; NKX2.1/TTF1: thyroid transcription factor 1; TTF2: thyroid transcription factor 2; PAX8: paied box gene 8; NKX2.5: NK2 homeobox 5; MCT8: monocarboxylate transporter 8; HC: hipotiroidismo congénito.
Cuando las concentraciones de hormonas tiroideas se normalizan durante el seguimiento se habla de hipotiroidismo congénito transitorio, cuya frecuencia es de entre 1:11,000-12,000. Puede ser secundario a la ingesta materna de fármacos antitiroideos, al paso transplacentario de anticuerpos antitroideos, a la deficiencia o exceso de yodo, a mutaciones heterocigotas de DUOX2 o DUOXA2 o a hemangiomas hepáticos congénitos que expresan niveles elevados de desyodasa tipo 3. También se presenta frecuentemente en recién nacidos con síndrome de Down. Se puede presentar hipotiroidismo transitorio de tipo central (niveles normales o bajos de la hormona estimulante de tiroides) en casos de hipertiroidismo materno, prematurez o en recién nacidos en estado crítico, particularmente si reciben dopamina, esteroides, aminofilina o cafeína7.
4Cuadro clínicoLa mayoría de los pacientes con HC no presentan datos clínicos al nacimiento. La fontanela posterior amplia (diámetro mayor a 0.5cm) es uno de los hallazgos más frecuentes. Otros datos que se presentan si no se inicia un tratamiento oportuno son macroglosia, edema, llanto ronco, facies tosca, hernia umbilical, hipotonía, piel moteada, hipotermia, letargia, ictericia prolongada (más de dos semanas), bradicardia, dificultad para alimentarse y estreñimiento. En ocasiones, el nacimiento es postérmino. La presencia de datos clínicos al nacimiento y un núcleo de osificación distal del fémur, ausente o menor de 3mm de diámetro, sugiere que el hipotiroidismo es severo y tanto materno como fetal. Es importante explorar la tiroides, ya que en caso de disgenesia generalmente no es palpable, y en caso de dishormogénesis se encuentra bocio.
Los pacientes con hipotiroidismo congénito presentan mayor prevalencia de hipoacusia y de malformaciones congénitas extratiroideas que la población general (8.4-10% vs. 3% en la población general)8,9, y sobre todo anomalías cardiacas (1.5-5.8%), paladar hendido y displasia de cadera (1.1-3.8%), así como malformaciones neurológicas, genitourinarias, digestivas y oftalmológicas.
5Diagnóstico5.1Tamiz neonatalDebido a los escasos datos clínicos al nacer y a la necesidad de iniciar tratamiento temprano para evitar secuelas, el HC es una enfermedad que debe buscarse mediante el tamiz neonatal. Existen varias estrategias que a continuación se enumeran:
- 1
La medición primaria de tetrayodotironina (T4) y la confirmación con la medición de la hormona estimulante de tiroides (TSH)
- 2
La medición primaria de TSH con la confirmación con
- 3
La medición primaria simultánea de T4 y TSH
En México se utiliza la segunda estrategia (medición primaria de TSH), ya que tiene las ventajas de ser de bajo costo, ser muy sensible y detectar hipotiroidismo subclínico. La desventaja que presenta es que no detecta hipotiroidismo de origen central ni casos de elevación tardía de TSH.
Cada programa de tamizaje para HC ajusta los puntos de corte de las concentraciones de TSH para maximizar la sensibilidad y minimizar la tasa de “re-llamado”. Se han propuesto niveles de TSH tan bajos como 6 mU/l y hasta 20 mU/l como puntos de corte10. En México, se toma la muestra para el tamiz —por punción y goteo de talón, impregnando un papel filtro— entre el segundo y quinto días de vida. Las mediciones antes de las 48h aumentan la frecuencia de falsos positivos, y las mediciones tardías aumentan el riesgo de retraso en el inicio del tratamiento. El resultado del tamiz neonatal debe ser comunicado antes de los 15 días de vida. Algunos falsos negativos ocurren por elevación tardía de TSH. Para disminuir el riesgo de subdiagnóstico en los casos de HC con elevación tardía de TSH, algunos programas, incluido el mexicano, indican la repetición del tamiz entre las 2 y 6 semanas de vida en los prematuros, en los pacientes que ingresaron a una unidad de cuidados intensivos neonatales, en pacientes con anomalías cardiovasculares y en gemelos monocigóticos. La medición de T4 diagnostica del 12 al 15% más de casos que son de origen central. Una concentración de TSH mayor de 40 mU/l o mayor de 20 mU/l acompañada de T4 menor de 5μg/dl son 100% específicos para el diagnóstico de HC permanente y ameritan el inicio urgente del tratamiento. De los casos con TSH entre 20 y 39 mU/l en el tamiz, el 75% son falsos positivos o casos de hipotiroidismo transitorio. Los casos de HC no detectados con los programas de tamiz neonatal pueden alcanzar hasta el 5%, lo cual, generalmente, se debe a errores humanos en el manejo de la muestra, el análisis o el reporte de los resultados.
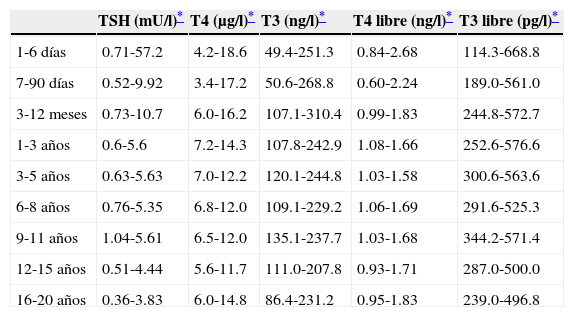
5.2Diagnóstico confirmatorioEl punto de corte para solicitar la prueba confirmatoria es una concentración de TSH de 10 mU/l determinada por fluoroinmunoensayo o por ELISA11. Para confirmar el diagnóstico, es necesario medir TSH y T4 total y/o libre en suero de sangre venosa en las siguientes 24h de comunicado el resultado del tamiz. La TSH mayor de 40 mU/l con T4 baja es indicativa de HC, que por lo general es permanente. Los pacientes con anomalías más leves pueden ser seguidos por muestreo repetido, ya que frecuentemente la alteración es transitoria (fig. 1). Es importante considerar los valores de referencia de hormonas tiroideas específicos para cada edad (tabla 2).
.")
Diagrama de flujo para el tamiz neonatal. PFT: pruebas de función tiroidea; TSH: hormona estimulante de la tiroides; T4 L: tetrayodotironina libre; HCC: hipotiroidismo congénito central.
1 Micropene, criptorquida, hipoglucemia, defectos de la línea media.
2 La gammagrafía se puede realizar hasta 5 días después de haber iniciado tratamiento, pero no se debe retrasar el mismo en espera de los estudios de imagen.
3 En estos casos se deja que el clínico tome la decisión de iniciar el tratamiento de inmediato y revalorar a los 3 años, o postergar la decisión tres semanas con nueva medición de hormonas tiroideas y estudios de imagen.
Basado en el Consenso de la Sociedad Europea de Endocrinología Pediátrica para el Tamizaje, Diagnóstico y Tratamiento del Hipotiroidismo Congénito (Léger y colaboradores, 2014).
Rangos de referencia para hormonas tiroideas según la edad
| TSH (mU/l)* | T4 (μg/l)* | T3 (ng/l)* | T4 libre (ng/l)* | T3 libre (pg/l)* | |
|---|---|---|---|---|---|
| 1-6 días | 0.71-57.2 | 4.2-18.6 | 49.4-251.3 | 0.84-2.68 | 114.3-668.8 |
| 7-90 días | 0.52-9.92 | 3.4-17.2 | 50.6-268.8 | 0.60-2.24 | 189.0-561.0 |
| 3-12 meses | 0.73-10.7 | 6.0-16.2 | 107.1-310.4 | 0.99-1.83 | 244.8-572.7 |
| 1-3 años | 0.6-5.6 | 7.2-14.3 | 107.8-242.9 | 1.08-1.66 | 252.6-576.6 |
| 3-5 años | 0.63-5.63 | 7.0-12.2 | 120.1-244.8 | 1.03-1.58 | 300.6-563.6 |
| 6-8 años | 0.76-5.35 | 6.8-12.0 | 109.1-229.2 | 1.06-1.69 | 291.6-525.3 |
| 9-11 años | 1.04-5.61 | 6.5-12.0 | 135.1-237.7 | 1.03-1.68 | 344.2-571.4 |
| 12-15 años | 0.51-4.44 | 5.6-11.7 | 111.0-207.8 | 0.93-1.71 | 287.0-500.0 |
| 16-20 años | 0.36-3.83 | 6.0-14.8 | 86.4-231.2 | 0.95-1.83 | 239.0-496.8 |
TSH: hormona estimulante de la tiroides; T4: tetrayodotironina; T3: triyodotironina.
Metodología: ELISA-electroquimioluminiscencia (modificado de Kratzch y colaboradores; 2008).
Si bien el tratamiento de HC no debe retrasarse hasta los estudios de imagen, la gammagrafía y el ultrasonido son de utilidad para conocer su etiología (fig. 2). El Tc99 es el radiofármaco de elección para identificar la localización de la tiroides, pues su depuración es más rápida y la exposición a la radiación es menor que con I123. La gammagrafía con I123 es necesaria cuando se desea cuantificar la captación tiroidea de yodo en casos de sospecha de dishormogénesis. La falta de captación del radiofármaco por la tiroides casi siempre indica aplasia (atirosis). Si no se observa la tiroides en la gammagrafía pero sí en el ultrasonido, las causas posibles son un defecto del receptor de TSH, un defecto en el transporte de yodo, anticuerpos bloqueadores de receptor de TSH o TSH baja por tratamiento con levotiroxina. La gammagrafía también puede demostrar una localización ectópica de la tiroides. La captación aumentada de I123 que disminuye a las 24 h respecto a la captación a las 4h sugiere dishormogénesis. Si han transcurrido más de 5 días de iniciado el tratamiento, la gammagrafía no es de utilidad puesto que la captación del radiofármaco será baja debido a los niveles bajos de TSH. El ultrasonido es poco sensible para identificar disgenesia tiroidea. Es posible realizar el diagnóstico molecular de las causas de dishormogénesis y de los hipotiroidismos sindrómicos cuando son debidas a mutaciones conocidas.

Diagnóstico etiológico de hipotiroidismo congénito primario.
1 El perclorato no está disponible en nuestro medio, pero una captación de I123 que disminuye a las 24 horas en comparación con la captación a las 4 horas orienta a dishormogénesis.
2 NIS: cotransportador de sodio/yodo.
3 TSH: hormona estimulante de la tiroides; LT4: levotiroxina. Puede existir captación baja.
Otros estudios diagnóstico que pueden ser informativos son la medición de tiroglobulina, de anticuerpos antitiroideos, la yoduria y la edad ósea. La concentración alta de tiroglobulina sugiere dishormogénesis mientras que una concentración muy baja o indetectable sugiere agenesia tiroidea o bien algún trastorno en la síntesis de esta proteína, aunque las concentraciones encontradas en estos pacientes se imbrican con los intervalos de normalidad. El hallazgo de anticuerpos antitiroideos en suero sugiere un proceso aloinmune que generalmente es transitorio. Es posible diagnosticar deficiencia o exceso de yodo mediante la medición de yoduria de 24h, que en neonatos debe ser de 50 a 100μg. La ausencia del núcleo de osificación de la epífisis distal de fémur (signo de la “rodilla vacía”) o un diámetro de este<3mm en un recién nacido de término sugiere hipotiroidismo severo de inicio intrauterino.
La utilidad del diagnóstico etiológico radica en el pronóstico. Si se confirma disgenesia tiroidea (ectopia o agenesia) o dishormogénesis, el hipotiroidismo es permanente sin duda. Si la etiología no se esclarece, ya sea por imposibilidad de realizar una gammagrafía antes de iniciar el tratamiento o por resultados no concluyentes de esta, en particular cuando la elevación de TSH es moderada (menor de 40 mU/l), se debe esperar hasta los tres años de edad para suspender el tratamiento y reevaluar, ya que la suspensión de tratamiento antes de esta edad en un niño con hipotiroidismo perjudicaría su neurodesarrollo.
El hipotiroidismo de tipo central (TSH normal o baja con T4 baja) no se identifica por el tamiz basado en la medición primaria de TSH. Hay que sospecharlo siempre que existan defectos de la línea media o datos de deficiencia de alguna otra hormona hipofisiaria, como hipoglucemia, micropene, criptorquidia o ictericia prolongada. En los pacientes con HC debe descartarse hipoacusia mediante pruebas audiológicas apropiadas para la edad, y evaluar la pertinencia de realizar estudios de gabinete para descartar malformaciones congénitas.
6TratamientoLa levotiroxina (LT4) es el tratamiento de elección para el HC. El objetivo es alcanzar un neurodesarrollo y crecimiento correspondientes al potencial genético del niño. Para esto, se debe iniciar con una dosis adecuada de LT4 dentro de las dos primeras semanas de vida. Si la concentración de TSH en el tamiz es mayor de 40 mU/l, el médico de primer contacto debe indicar LT4 a la brevedad y sin esperar la valoración especializada ni el resultado de la prueba confirmatoria (aunque siempre que sea posible debe tomarse muestra venosa para determinación de TSH y T4 libre antes de empezar el tratamiento). Si la concentración de TSH es elevada pero menor de 40 mU/l, se puede esperar un par de días al resultado del análisis de sangre venosa para iniciar el tratamiento; si dicho análisis revela T4 libre baja para la edad o TSH mayor a 20 mU/l, aunque la T4 libre sea normal, se debe iniciar el tratamiento. Si la T4 libre es normal y la TSH se encuentra entre 6 y 20 mU/l después de los 21 días de vida, puede optarse por esperar el resultado de estudios de imagen con vigilancia estrecha y repetición de perfil tiroideo dos semanas después, o iniciar el tratamiento inmediato y reevaluar hasta los 3 años de edad (fig. 1).12
Las metas bioquímicas del tratamiento con levotiroxina son las siguientes:
- 1)
Normalizar la concentración de T4 libre lo más pronto posible, es decir, en la primera semana de iniciada la sustitución
- 2)
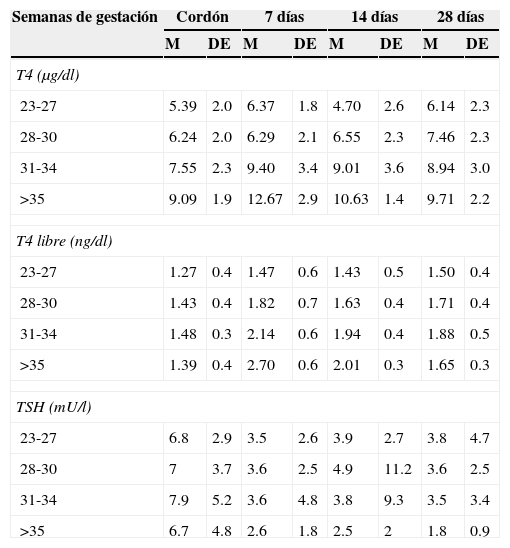
Mantener la T4 libre en la mitad superior de los rangos de referencia acordes con la edad (tabla 3)13
Tabla 3.Rangos de referencia para hormonas tiroideas en recién nacidos según edad gestacional
Semanas de gestación Cordón 7 días 14 días 28 días M DE M DE M DE M DE T4 (μg/dl) 23-27 5.39 2.0 6.37 1.8 4.70 2.6 6.14 2.3 28-30 6.24 2.0 6.29 2.1 6.55 2.3 7.46 2.3 31-34 7.55 2.3 9.40 3.4 9.01 3.6 8.94 3.0 >35 9.09 1.9 12.67 2.9 10.63 1.4 9.71 2.2 T4 libre (ng/dl) 23-27 1.27 0.4 1.47 0.6 1.43 0.5 1.50 0.4 28-30 1.43 0.4 1.82 0.7 1.63 0.4 1.71 0.4 31-34 1.48 0.3 2.14 0.6 1.94 0.4 1.88 0.5 >35 1.39 0.4 2.70 0.6 2.01 0.3 1.65 0.3 TSH (mU/l) 23-27 6.8 2.9 3.5 2.6 3.9 2.7 3.8 4.7 28-30 7 3.7 3.6 2.5 4.9 11.2 3.6 2.5 31-34 7.9 5.2 3.6 4.8 3.8 9.3 3.5 3.4 >35 6.7 4.8 2.6 1.8 2.5 2 1.8 0.9 M: media; DE: desviación estándar; T4: tetrayodotironina; T4 L: tetrayodotironina libre; TSH: hormona estimulante de la tiroides.
Valores tomados en neonatos con diversas patologías.
Metodología: T4 y TSH, radioinmunoanálisis; T4 libre, quimioluminiscencia (modificado de Williams y colaboradores; 2004).
- 3)
Que la concentración de TSH se mantenga entre 0.5 y 2 mU/l a partir del primer mes de tratamiento14
Para lograr estas metas, la dosis diaria de levotiroxina debe ser de 10 a 15μg/kg13,15. Generalmente se inicia con una dosis aproximada de 50μg diarios, aunque después sea necesario disminuirla. Es necesario verificar los niveles séricos de T4 total y/o libre y de TSH entre una y dos semanas después de iniciado el tratamiento.
La levotiroxina debe administrarse en forma de tabletas molidas y suspendidas en algunos mililitros de agua mediante una cuchara pequeña de metal. No se debe intentar diluir ni administrar en algún dispositivo de plástico, como jeringa o biberón, ya que no es hidrosoluble y se adhiere a las superficies de plástico. El hierro, la soya, la fibra, el sucralfato, la colestiramina, el calcio y el hidróxido de aluminio interfieren con la absorción de levotiroxina. Esta debe ser administrada en ayuno (al menos 30 minutos antes del primer alimento) o con leche materna, pero siempre de la misma forma. Es necesario concientizar a los padres y cuidadores sobe la importancia de la adherencia estricta al tratamiento, entregarles las indicaciones detalladas por escrito y administrar la primera dosis en el consultorio. Recientemente, en algunos países europeos, se encuentra disponible una formulación líquida más fácil de administrar a los lactantes que las tabletas (Tirosint gotas, IBSA Farmaceutici, Italia). Estudios preliminares parecen demostrar una adecuada absorción, aparentemente sin diferencias en el neurodesarrollo y crecimiento respecto del tratamiento convencional. Sin embargo, existen reservas sobre el contenido de etanol y la estabilidad de esta formulación16. No se debe administrar una formulación líquida de LT4 no autorizada y cuya farmacocinética no se haya evaluado apropiadamente. La administración simultánea de triyodotironina (T3) no tiene ninguna utilidad ya que la mayoría de la T3 en el sistema nervioso central se produce por monodesyodación local de T4.
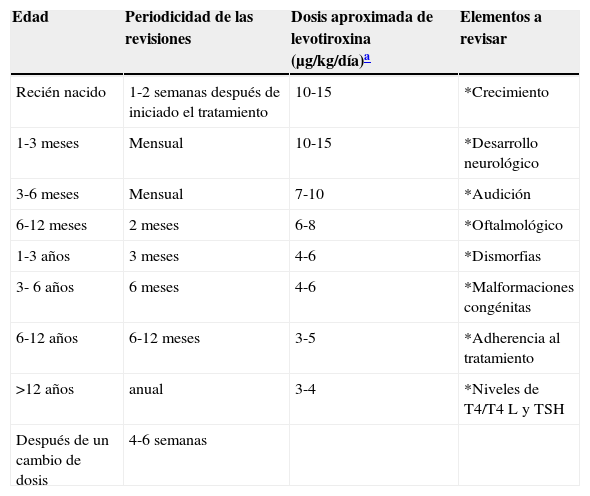
En la tabla 4 se enlistan las recomendaciones para el seguimiento de los pacientes. Además de verificar las concentraciones séricas de hormonas tiroideas, es importante descartar hipoacusia, alteraciones visuales y malformaciones congénitas durante el seguimiento, vigilar el crecimiento y el neurodesarrollo. Conviene iniciar tempranamente con la terapia de neurorehabilitación. Es necesario verificar los niveles de hormonas tiroideas 4 semanas después de un cambio en la dosis. En algunos casos la TSH no se normaliza aun con niveles adecuados de T4. En estos casos es importante descartar un mal apego al tratamiento o una mala técnica de administración, aunque en algunos pacientes puede ser necesario llegar a niveles suprafisiológicos de T4 para mantener la TSH por debajo de 2 mU/l. El sobretratamiento se ha asociado con hiperactividad, agresividad y craneosinostosis17.
Recomendaciones para el seguimiento de pacientes
| Edad | Periodicidad de las revisiones | Dosis aproximada de levotiroxina (μg/kg/día)a | Elementos a revisar |
|---|---|---|---|
| Recién nacido | 1-2 semanas después de iniciado el tratamiento | 10-15 | *Crecimiento |
| 1-3 meses | Mensual | 10-15 | *Desarrollo neurológico |
| 3-6 meses | Mensual | 7-10 | *Audición |
| 6-12 meses | 2 meses | 6-8 | *Oftalmológico |
| 1-3 años | 3 meses | 4-6 | *Dismorfias |
| 3- 6 años | 6 meses | 4-6 | *Malformaciones congénitas |
| 6-12 años | 6-12 meses | 3-5 | *Adherencia al tratamiento |
| >12 años | anual | 3-4 | *Niveles de T4/T4 L y TSH |
| Después de un cambio de dosis | 4-6 semanas |
T4: tetrayodotironina; T4 L: tetrayodotironina libre; TSH: hormona estimulante de la tiroides.
El efecto deletéreo del hipotiroidismo en el neurodesarrollo de niños menores de 3 años está ampliamente estudiado. El retraso de tan solo una semana en el inicio del tratamiento sustitutivo tiene efectos negativos en la cognición18. Son factores pronóstico para un neurodesarrollo subóptimo la atirosis, la edad ósea retrasada (núcleo distal de fémur < 0.5 cm2 en un neonato de término), niveles de T4<2μg/l al momento del diagnóstico, lapso de tiempo prolongado para normalizar la TSH, inicio de tratamiento después de la segunda semana de vida y cuatro o más episodios de TSH >5 mU/l en los primeros 3 años de vida13,19–21.
Recientemente se ha cuestionado si los niños que reciben el tratamiento recomendado tienen un neurodesarrollo óptimo. Se ha reportado que estos niños pueden tener alteraciones neurocognitivas leves; algunos estudios reportan un coeficiente intelectual de 5 a 25 puntos más bajo que los niños sanos22, mientras que otros no encuentran diferencias, o solo las identifican en algunas áreas específicas, como atención y audición17,18. En cuanto al hipotiroidismo de etiología transitoria, el efecto de los fármacos antitiroideos persiste hasta las 2 o 3 semanas de vida postnatal, y generalmente no requiere tratamiento. El HC por presencia de anticuerpos antitiroideos suele resolverse a los 2-3 meses de edad y generalmente sí requiere de sustitución con hormonas tiroideas durante este periodo23.
8Casos especiales8.1Hipertirotropinemia aislada (TSH alta y T4 normal)La etiología es heterogénea e incluye la mutación en el receptor de TSH, la deficiencia de yodo y el síndrome de Down, aunque frecuentemente es idiopática y algunas veces la elevación de TSH es transitoria. Existen controversias respecto al tratamiento; en general se recomienda repetir las mediciones dos semanas después, y dar tratamiento si la TSH persiste mayor a 10 mU/l o la concentración de T4 libre disminuye13.
8.2Hipotiroxinemia aislada (T4 baja y TSH normal)Este patrón suele observarse en prematuros, neonatos enfermos o en quienes se administra dopamina o glucocorticoides. Si bien se ha descrito una asociación entre hipotiroxinemia y un pronóstico neurológico adverso, no está claro si la asociación es causal, y no hay consenso respecto al tratamiento de estos pacientes. Si la T4 es baja, pero la T4 libre y TSH son normales, el diagnóstico más probable es deficiencia de proteína lijadora de tiroxina (TBG), y no se requiere tratamiento. El hipotiroidismo secundario o terciario, es decir, de origen central, también puede presentarse como hipotiroxinemia aislada. Por último, existen pacientes con hipotiroidismo primario con elevación tardía de TSH, es decir, posterior a la fecha de toma del tamiz, y este patrón es más frecuente en prematuros y neonatos en estado crítico24,25.
8.3Disfunción tiroidea en el paciente con síndrome de DownLos pacientes con síndrome de Down tienen una prevalencia aumentada hasta 30 veces mayor que la población general, tanto de HC (1.5-6.1%) como de disfunción tiroidea adquirida26. También existe mayor frecuencia de hipertirotropinemia aislada o hipotiroidismo subclínico (25.3-60%) y de autoinmunidad tiroidea (7.5-34%). Entre 40 y 80% tiene elevaciones aisladas y leves de TSH27, aunque aparentemente en la mayoría de los casos esta elevación es transitoria o no evoluciona a hipotiroidismo franco28. Se han propuesto diversos mecanismos para explicar la disfunción tiroidea de los pacientes con síndrome de Down. Entre estos se encuentran la existencia de disfunciones del eje hipotálamo-tiroides, insensibilidad leve a TSH y bioactividad disminuida de TSH, sin que se haya corroborado ninguno de ellos28. El hipotiroidismo podría agravar el déficit cognitivo, el crecimiento y los factores de riesgo cardiometabólico presentes en los pacientes con síndrome de Down29. Sin embargo, no es claro si la hipetirotropinemia aislada o hipotiroidismo subclínico tiene un efecto nocivo en estos pacientes30. Se ha descrito que los pacientes con síndrome de Down e hipertirotropinemia o T4 libre por debajo de la mediana tienen menores niveles de hemoglobina, mayor prevalencia a anemia y se encuentran más hipotónicos que los que tienen TSH normal y T4 libre por arriba de la mediana31. Se recomienda realizar un perfil tiroideo al nacimiento, a los 6 y 12 meses de vida, y posteriormente una vez al año en los sujetos con síndrome de Down. Sin embargo, no es claro cuándo un paciente con hipertirotropinemia aislada debe recibir tratamiento ya que en muchas ocasiones esta es transitoria32. Se ha sugerido que se inicie tratamiento si la TSH es mayor de 10 mU/l, si hay síntomas o si hay anticuerpos antiperoxidasa33.
8.4Disfunción tiroidea en recién nacidos con prematurezLa función tiroidea del recién nacido con prematurez es distinta respecto de la del recién nacido de término. Frecuentemente, los recién nacidos con prematurez presentan alteraciones en las pruebas de función tiroidea que son difíciles de interpretar. Las concentraciones de hormonas tiroideas varían de acuerdo con la edad postconcepción y con los días de vida extrauterina34 (tabla 3). Sin embargo, es difícil establecer valores de “normalidad” pues los neonatos prematuros casi siempre cursan con múltiples comorbilidades y reciben múltiples fármacos que interfieren con la función tiroidea. El hipotiroidismo congénito en prematuros es más frecuente que en niños de término; a pesar de ello, este puede no ser diagnosticado en el primer tamiz neonatal porque, generalmente, en<1% de los menores de 1,500g35 se encuentra “elevación tardía de TSH” y porque el pico de TSH en los prematuros no es tan acentuado como en los niños de término23. Hasta el 30% de los casos de elevación tardía de TSH pueden corresponder a hipotiroidismo permanente. Por lo anterior, es importante repetir el tamiz neonatal a las 4-6 semanas de vida y antes del egreso, para evitar falsos negativos en estos pacientes.
Los prematuros son más sensibles al exceso de yodo —por exposición a fármacos como medios de contraste o antisépticos—, además de que la absorción por la piel es mayor y la depuración es menor que en niños de término. También son más susceptibles a la deficiencia de yodo, ya que sus reservas son más bajas y sus requerimientos más altos que en niños de término, y no son cubiertos por la leche materna ni las fórmulas infantiles ni la nutrición parenteral. Hasta el 50% de los neonatos menores de 28 semanas de gestación cursan con “hipotiroxinemia transitoria”36, es decir, T4 libre baja con TSH normal. Es posible que este fenómeno se deba a una inmadurez del eje hipotálamo-hipófisis-tiroides y/o tenga mecanismos análogos a la disfunción tiroidea del paciente crítico o síndrome del eutiroideo enfermo. La hipotiroxinemia se asocia con desenlaces adversos en prematuros37, pero no se sabe si el tratamiento sustitutivo les confiere algún beneficio25,38.
FinanciaciónNinguna.
Conflicto de interesesLa autora declara no tener ningún conflicto de intereses.