






A glass-ceramic in the CaO–P2O5–SiO2 system, which contains two polymorphic modifications of tricalcium phosphate – whitlockite, β-Ca3(PO4)2 and α-Ca3(PO4)2, has been synthesized by the sol–gel method and thermal treatment up to 1200°C. The phase composition and microstructure of the glass-ceramic were investigated with X-ray diffraction analysis (XRD), Fourier-transformed infrared spectroscopy (FTIR), scanning electron microscopy and energy dispersive spectroscopy (SEM-EDS). An in vitro bioactivity test of the glass-ceramic in a simulated body fluid (SBF) was conducted for up to 21 days. α-Ca3(PO4) dissolved almost completely in SBF after 7 days. The experimental results of XRD, FTIR, SEM and EDS clearly validated the ability of the glass-ceramic samples to form a layer of hydroxyapatite on their surface in an SBF environment. We also studied the cytotoxic effect of the glass-ceramic on murine bone marrow (BM) cells and pre-osteoclasts in vitro. The glass-ceramic reduced the viability of BM cells in a dose-dependent manner being less toxic at concentrations below 0.1mg/ml. It modestly affected the viability of pre-osteoclasts cultured in osteoclast differentiation media. The obtained sample increased the percentage of pre-osteoclasts expressing the receptors involved in osteoclastogenesis (RANK) and apoptosis (TRAIL). In conclusion, the glass-ceramic showed the potential to affect the survival/differentiation of pre-osteoclasts at early stage of osteoclastogenesis. It might be suitable for tissue engineering including implants coating or scaffold as it can interfere with early stage of osteoclastogenesis which is required for proper bone remodelling and repair upon a long-term application of biomaterials.
Una vitrocerámica en el sistema CaO-P2O5-SiO2, que contiene dos modificaciones polimórficas de fosfato tricálcico: whitlockita, β-Ca3(PO4)2 y α-Ca3(PO4)2, ha sido sintetizada por el método sol-gel y tratamiento térmico hasta 1.200°C. La composición de fase y la microestructura de la vitrocerámica se investigaron por análisis de difracción de rayos X (XRD), espectroscopia infrarroja por transformada de Fourier (FTIR), microscopia electrónica de barrido y espectroscopia por dispersión de energía de rayos X (SEM-EDS). Se realizó una prueba de la bioactividad in vitro de la vitrocerámica por inmersión en un fluido corporal simulado (SBF) durante 21 días. α-Ca3(PO4) se disolvió casi completamente en SBF después de 7 días. Los resultados experimentales de XRD, FTIR, SEM y EDS validaron claramente la capacidad de las muestras de vitrocerámica para formar una capa de hidroxiapatita en su superficie en un entorno SBF. También estudiamos el efecto citotóxico de la vitrocerámica en células de médula ósea murina (MO) y preosteoclastos in vitro. La vitrocerámica redujo la viabilidad de las células de MO de forma dosis-dependiente, siendo menos tóxica a concentraciones inferiores a 0,1mg/ml. Afectó modestamente a la viabilidad de los preosteoclastos cultivados en medios de diferenciación de osteoclastos. La vitrocerámica aumentó el porcentaje de preosteoclastos que expresan los receptores implicados en la osteoclastogénesis (RANK) y la apoptosis (TRAIL). En conclusión, la vitrocerámica mostró el potencial de afectar la supervivencia/diferenciación de los preosteoclastos en la etapa temprana de la osteoclastogénesis. Podría ser adecuada para la ingeniería de tejidos, incluidos los recubrimientos de implantes o andamios, ya que puede interferir con la etapa temprana de la osteoclastogénesis que se requiere para la remodelación y la reparación ósea adecuada tras la aplicación a largo plazo de biomateriales.
The term biphasic calcium phosphate (BCP) was first used by Nery et al. [1] to describe the bioceramic that consisted of a mixture of hydroxyapatite (HA) and β-tricalcium phosphate (β-TCP), based on XRD analysis, as his co-worker LeGeros reported [2,3]. The BCP was first prepared in 1986 as a mixture of HA and β-TCP [4]. This new ceramic composition is characterized by the optimal balance of the more stable hydroxyapatite (HA) and more soluble α-tricalcium phosphate (α-TCP) and/or β-TCP [5,6]. The obtained final HA/β-TCP or HA/α-TCP materials are soluble and gradually dissolve in different fluids, seeding new bone formation as they release Ca2+ and PO43− ions into the biological medium [4,5].
From a chemical point of view, HA is stable when sintered below 1200°C, but beyond 1200°C HA loses its OH groups gradually and transforms to oxyapatite Ca10(PO4)6O. At 1450°C, oxyapatite is decomposed to α/-TCP, Ca2P2O7 and Ca4P2O9[7]. HA is slow resorbing at physiological conditions, whereas α-TCP and β-TCP are more soluble and hence show higher bioactivity [4,5,8]. On the other hand, there are three polymorphs of TCP, one the low-temperature β-TCP [6,9,10], and two high-temperature forms, α-TCP [9,10] and α/-TCP [9]. α/-TCP is of no practical interest because it only exists at temperatures>1430°C and reverts almost instantly to α-TCP when cooled below the transition temperature [6,9]. In contrast, β-TCP is stable at room temperature and transforms reconstructively at 1125°C to α-TCP, which can be retained during cooling to room temperature [11].
BCP is obtained when calcium-deficient hydroxyapatite (CDHA), with chemical formula Ca10−xMx(PO4)6−y(HPO4)y(OH)2[2], is sintered at a temperature above 700°C [4–6,9,10,12]. Some authors concluded, that after thermal treatment, CDHA transformed to HA/α-TCP or β-TCP [12].
Pure β-TCP, pure HA and BCP (HA/β-TCP) samples were also synthesized from different precursors at different Ca/P molar ratio [13,14].
Studies demonstrating the efficacy of HA, α-TCP, β-TCP, and BCP in tissue engineering have expanded alongside with the increase in interest in the techniques for synthesis of HA, α-TCP, β-TCP, and BCP, reviewed by Bal et al. [15]. These materials resemble the inorganic bone and thus can be used to repair bone defects and degenerative failures. They are a good alternative to auto- and allografts as they are biodegradable, biocompatible, and bioactive materials and are widely used as a scaffold for bone regeneration [16]. However, there is still a need for research on the biological performance and structural characteristics of these densified glass-ceramic materials, formation of HA on their surface in the presence of Mg2+, Si4+ ions in the environment.
In our previous work, we synthesized new glass-ceramic material via the sol–gel method in the system CaO–SiO2–P2O5 from CaO, TEOS and H3PO4 in the presence of HCl acid as a catalyst [17]. The sol–gel method is widely applied in the synthesis of bioglass and bioceramics. It allows influencing the phase composition and structure of the obtained materials, through a number of factors such as precursors, pH, etc. In addition to the high degree of homogeneity and the possibility of synthesis at low temperatures, it is assumed that it also may have a different course of phase formation compared to precipitation, solid-state sintering, crystallization of glass, crystallization in a melt, etc. The obtained BCP (HA/β-TCP) powder was used as a filler to prepare natural rubber composites applicable to flexible antennae [17].
In this context the present article intends (1) to investigate the possibility of obtaining a new sintered TCP silicate glass-ceramic by sol–gel method; (2) to establish the phase composition and microstructure of the obtained sample and to identify the sample changes in a SBF solution, as a function of the sample's immersion time in it.; (3) to demonstrate the potential of the TCP silicate glass-ceramic for implication in tissue engineering. The obtained experimental data are expected to provide useful information that will allow, by means of the synthesis conditions and control of the phase composition and microstructure, to obtain biomaterials with specified properties.
In order to demonstrate the potential of the α/β-TCP silicate glass-ceramic for implication in tissue engineering we study the cytotoxicity of the TCP on murine bone marrow cells and pre-osteoclasts as well as we delineate its impact on the surface expression of receptors involved in osteoclast differentiation, like the receptor activator of nuclear factor kappa-B (RANK) and in apoptosis, like the surface form of the tumour necrosis factor-related apoptosis-inducing ligand (TRAIL).
Experimental procedureSynthesis of the TCPThe materials obtained were synthesized by a poly-step sol–gel method. The first step was to prepare SiO2 sol from TEOS (Si(OC2H5)4). TEOS was stirred in the solvent composed of C2H5OH and H2O with a very small amount of HCl, as a catalyst, in a volume ratio of TEOS:C2H5OH:H2O:HCl=1:1:1:0.01. When the solution of the described mixture became transparent in approximately 1h, a mixture of calcium and phosphate sources was added under intensive stirring. The calcium phosphate solution was prepared by mixing Ca(OH)2 and H3PO4 at pH=10. Calcium phosphate solution was added to SiO2 sol under stirring for 20h [39]. The sol obtained was gelated at 120°C for 12h and then thermally treated at 1200°C for 2h. The thermal treatment of the dry gel previously homogenized in an agate mortar was carried out in a corundum crucible. It is noteworthy that without pressing the starting powder material, sintering and densification processes took place with the participation of a liquid phase (melt), during which the volume of the starting material was significantly reduced and a strong, compact and dense material was obtained. The chemical composition of the prepared glass-ceramic CaO–P2O5–SiO2 was 58.2 CaO, 29.4 P2O5, 12.5 SiO2 (wt.%), where Ca/P+Si=1.7 (mol. ratio).
Characterization of the TCP by X-ray powder diffraction (XRD)X-ray powder diffraction (XRD) analysis was applied for phase control. An X-ray diffractometer Empyrean (PANalytical) at CuKα radiation was used in the range from 8° to 80° 2θ (step size: 0.05°, counting time per step: 38.5s). The crystalline phases were identified using the powder diffraction files PDF # 70-2065, PDF # 29-0359 and PDF # 01-072-1243 from the database JCPDS-International Centre for Diffraction Data PCPDFWIN. The quantitative phase analysis of the samples and the determination of the unit cell parameters of the crystalline phases were performed by using the Powder Cell 2.4 software [18].
Characterization of the TCP by Fourier-transform infrared spectroscopy (FTIR)Fourier-transform infrared spectroscopy (FTIR) was applied by using the pressed-pellet technique in KBr. The measurements were done with a spectrophotometer Varian 600-IR series (spectral resolution not worse than 0.07cm−1) in the frequency range from 4000 to 400cm−1.
Characterization of the TCP by optical microscopy and scanning electron microscopy SEM–EDXObservations of the morphology of broken surfaces of the bulk glass-ceramic were carried out with a ZEISS Stemi 305 Stereo Microscope and with SEM. The contact surfaces of the prepared samples (pellets) after in vitro test were observed using Scanning Electron Microscopy on SEM/FIB LYRA I XMU (TESCAN) equipment with energy dispersive spectroscopy (EDS) facilities – Quantax 200 (Bruker) detector.
In vitro bioactivityTo estimate the in vitro bioactivity of the prepared sample, we used the Simulated Body Fluid (SBF) following different ions: Na+ 142.0, K+ 5.0, Mg2+ 1.5, Ca2+ 2.5, Cl− 148.8, HCO3− 4.2 and PO42− 1.0mM and buffered at 7.4 pH, proposed by Kokubo et al. [19]. The concentrations of various ions in the SBF were adjusted to be similar to those in human blood plasma. To obtain pellets, 0.5g of the homogenized glass-ceramic powders with a particle size between 80 and 100μm were pressed uniaxially at a 12MPa. They were 0.5mm in thickness and 10mm in diameter. Three pellets were placed in polyethylene bottles containing 50ml of SBF at 37±0.5°C. The sample surface area to the SBF volume (SA/V) ratio was equal to 0.1cm−1.
Pellets were taken out of the solution after the 3rd, 7th, 14th, and 21st day of soaking, gently rinsed with deionised water and acetone, and dried in air at room temperature. Sample surfaces before and after exposure to the SBF were examined by SEM and EDS. The changes in the samples during the in vitro test were examined by FTIR. The concentration of different ions in SBF solutions was determined by inductively coupled plasma optic emission spectrometry (Prodigy High Dispersion ICP-OES Spectrometer from Teledyne Leeman Labs).
Effect of the α/β-TCP on cell viability of murine bone marrow cells and pre-osteoclastsPreparation of the α/β-TCP for cell culture experimentsThe powder of α/β-TCP was measured by using analytical balance (ABS 220-4N, Kern, Stuttgart Germany), added in a sterile clear glass vial (#29372-U, Supelco®, Sigma–Aldrich, Munich, Germany) and immersed in sterile α-Minimum Essential Medium (α-MEM) with Eagle salts, l-glutamine and sodium bicarbonate, without sodium pyruvate (#M4655, Sigma–Aldrich, Munich, Germany) supplemented with 1× antibiotic/antimycotic solution (#A5955; 100× concentrated; (10,000U penicillin, 10mg streptomycin and 25μg amphotericin B per ml), Sigma–Aldrich, Munich, Germany) at a concentration of 200mg/ml at 37°C for 24h. After centrifugation at 800×g for 5min and filtration through a 0.22μm syringe filter (#SLGV013SL, PVDF membrane, Sigma–Aldrich, Germany), the supernatants were collected carefully. Subsequently, the supernatants were supplemented with 10% foetal bovine serum (FBS) (#F7524, Sigma–Aldrich, Germany). The extracts were stored at 4°C and used on the next day for culture experiments.
Isolation of murine bone marrow cells and generation of pre-osteoclastsBalb/c mice were purchased from Charles River Laboratories (Wilmington, MA, USA) and then bred in the Experimental Animals Facility at the Stephan Angeloff Institute of Microbiology (Sofia, Bulgaria). Mice (female or male, 6-week-old, weight 17–21g) were kept under standard conditions, and fed according to a laboratory diet (29% protein, 13% fat, 56% carbohydrates) and water ad libitum. The procedure was approved by National Food Agency (Sofia, Bulgaria) and executed according to the European Union Directive 2010/63/EU for animal experiments (License for Animal Housing No 352/30.01.2012 (registration No 11130005); License for Experimental Procedures No 105/10.07.2014). All experiments were conducted in accordance with the ARRIVE criteria (Animal Research: Reporting of In Vivo Experiments) and the principles of the 3Rs (Replacement, Reduction and Refinement) and under daily control by a certified veterinarian and the regular control by the representatives of the National Food Agency.
The femur and tibia of 6-week-old wild-type Balb/c mice (n=6) were aseptically collected. Bone marrow (BM) was isolated by flushing with 2ml α-MEM (supplemented with 1× antibiotic/antimycotic solution) using a sterile syringe with a 26G ¼ needle (KDM®, Berlin, Germany). BM suspension was prepared in a sterile petri dish by non-vigorous resuspending using a syringe with a 22G ¾ needle and then was centrifuged at 250×g for 10min, 4°C. The supernatant was discarded and the cells were washed two times with sterile phosphate buffer (PBS) containing 137mM NaCl, 2.7mM KCl, 10mM Na2HPO4, 1.8mM KH2PO4 (all molecular grade chemicals from Sigma–Aldrich, Munich, Germany) and resuspended in 10% FBS/α-MEM at concentration 2×106/ml. Half of the cells were seeded in a culture dish (#D7804, Nunclon®, Sigma–Aldrich, Munich, Germany) in the presence of 30ng/ml recombinant murine macrophage-colony stimulating factor (M-CSF) (#576404, BioLegend, London, UK). Non-attached BM monocytes (BMDMs) were collected after 48h, washed with PBS, counted and resuspended in fresh 10% FCS//α-MEM medium supplemented with 50ng/ml of recombinant murine receptor activator of nuclear factor kappa-B ligand (RANKL) (#577102, BioLegend, London, UK) and 30ng/ml M-CSF (#576404, BioLegend, London, UK). The generated cells were bone marrow-derived monocytes or pre-osteoclasts and were used in a proliferation assay or flow cytometry analyses.
Evaluation of cell viability by colorimetric assayCell viability was determined by MTT kit (#ab211091, Abcam, Cambridge, UK). The α/β-TCP at a solid/liquid ratio ranging from 0.0001 to 100mg/ml was added in a volume of 100μl/well to a flat bottom 96-well culture plate (#CLS351172, Corning-Falcon®, Sigma–Aldrich, Munich, Germany) and was let to disperse at the well bottom for 30min at 37°C in 5% CO2 incubator (IsoMed, Memert, Buechenbach, Germany). Then BM or pre-osteoclasts at a concentration of 5×104/ml in 10%FCS/α-MEM were carefully seeded in a volume of 100μl/well. The culture medium of pre-osteoclasts was supplemented with RANKL and M-CSF as described above. After 36h, the plates were spined at 550×g, 4°C for 10min (Hettich 320R® Centrifuge, Tuttlingen, Germany), 100μl of MTT reagent (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) dissolved 1:1 in media without FBS were added into each well. A control group contained 50μl MTT reagent and 50μl cell culture media without cells. The plate was incubated for 3h, at 37°C, and 5% CO2 and 150μl of MTT solvent were added into each well. The plate was incubated on an orbital shaker for 15min and the absorbance at optical density (OD) 590nm was measured by a microplate reader with Gen 5.0 software (Bioteck®, Agilent Technologies Santa Clara, CA, USA). The OD values for the control without cells were subtracted from each reading and the percentage of cytotoxicity was calculated using the following equation: % Cytotoxicity=(100×(Untreated cells−α/β-TCP-treated cells)/Untreated cells.
Evaluation of receptor expression by flow cytometryThe cell surface receptor expression was determined as previously described [20]. Briefly, BM or pre-osteoclasts cells (1×105cells/ml) in 2% BSA/PBS were subjected to Fc blocking for 10min on ice with anti-CD16/32 antibody (#101302; clone 93; 1.0μg/1×106cells/ml; BioLegend, London, UK). After washing with 2% BSA/PBS, the cells were incubated for 20min at 4°C, in dark, with validated concentrations of allophycocyanin (APC)-conjugated antibody against mouse Ly6C (clone HK1.4, #128015), phycoerythrin (PE)-conjugated antibody against TNF-related activation-induced cytokine (TRANCE, RANK) (clone R12-31; #119805) and phycoerythrin (PE)-labelled antibody against TNF-related apoptosis-inducing ligand (TRAIL; CD253) (clone RIK-2; #308205), and of the corresponding isotype rat control antibodies (all from BioLegend, London, UK). After washing, the samples were subjected to flow cytometry by the acquisition of at least 20,000cell counts/sample and live/dead cell discrimination by a BSR II flow cytometer (Becton Dickinson GmbH, San Jose, CA, USA). The data were obtained using the BD FACSDiva v6.1.2 Software (Becton Dickinson GmbH, San Jose, CA, USA) and analyzed by Cyflogic software 1.2.1 (CyFlo Ltd, Turku, Finland).
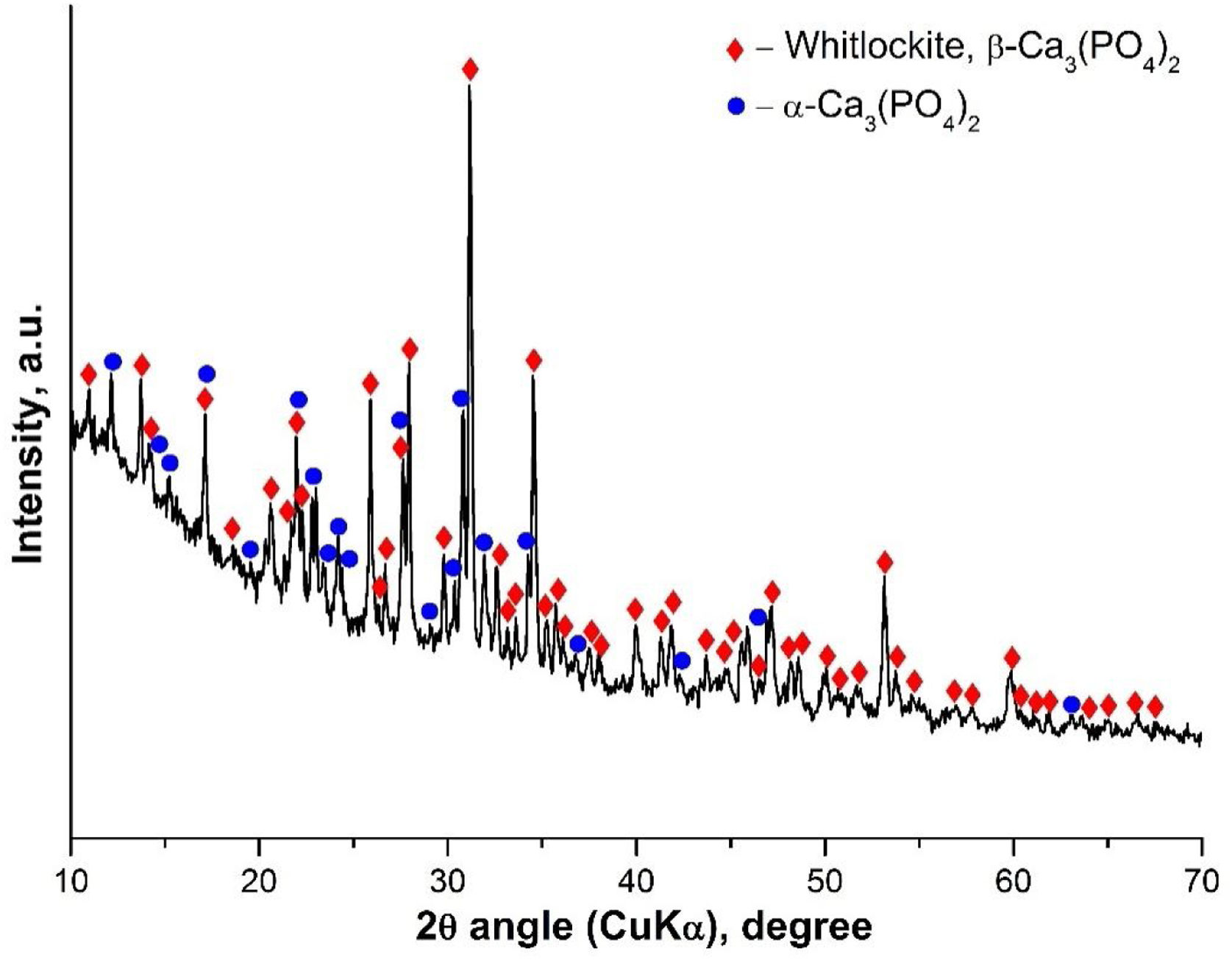
Results and discussionInitial characterization of the α/β-TCPThe α/β-TCP was synthesized by a poly-step sol–gel method as described above. The synthesized glass-ceramic is a bulk material with bulk density 2.63g/cm3, water absorption 0.79% and without macroscopically visible pores. Under magnification, spherical pores less than 1mm in diameter can be seen. In Fig. 1 a photograph of the fractured surface is shown. We then performed XRD analyses. The obtained X-ray diffraction pattern of the sample is shown in Fig. 2.


XRD results showed the presence of two crystalline phases: β-TCP (β-Ca3(PO4)2-structure type Whitlockite, with rhombohedral symmetry (PDF #70-2065) and α-TCP (α-Ca3(PO4)2 with monoclinic symmetry (PDF #29-0359). The amorphous halo between 10 and 25° 2θ in the diffraction pattern is due to a glassy phase formed during the final heat treatment. Using Powder Cell software (Rp=3.99, Rwp=5.57, Rexp=3.97) the quantitative ratio of the crystal phases β-TCP:α-TCP=46:54 was determined as well as the corresponding parameters of the unit cell for β-TCP: a=10.399Å, c=37.311Å, and for α-TCP: a=12.895Å, b=27.320Å, c=15.237Å, β=126.40°. The determined unit cell parameters for β-TCP are smaller, compared to the used structural data, in accordance with ICSD #97500, a=10.4352(2)Å and c=37.4029(5)Å. This result can be explained by the incorporation of Si4+ and Mg2+ in the structure, respectively replacing P5+ and Ca2+, and occupying their structural positions. The substitutions of Si4+ for P5+ and Mg2+ for Ca2+ in TCP have been the subject of a number of studies [21,22].
On the other hand, the substitution of Si4+and Mg2+ in TCP is due to their significance for living organisms. Both are essential for bone metabolism and bone formation. In our case, MgO was included as the impurity of the initial CaO precursor. The quantity of Mg2+ in our sample is less than 1wt%, which can be seen from EDX data, indicating that the quantity of Mg2+ in our sample is close to bone-like amounts of Mg. Mg in the β-TCP can increase the β→α transformation temperature and even small amounts of it, is able to postpone this transformation resulting in accelerated densification process during the sintering of β-TCP. Similarly, the presence of Si4+ and Mg2+ enhanced the thermal stability of β-TCP and α-TCP in solid TCP solution co-doped with the ions [22]. In this context, the presence of Mg in our α/β-TCP glass-ceramic, may improve the density properties of the material.
The presence of the α-TCP and β-TCP in the glass-ceramic material is compatible with the results obtained by other group [23] for the system Mg3(PO4)2–Ca3(PO4)2 particularly with the binary phase field where β-TCP and α-TCP solid solutions coexist. In our case, it is likely that the high-temperature transformation of HA will result in the formation of α-TCP. The glass-ceramic containing HA and β-TCP were synthesized similarly, after heat treatment at 1100°C according to our previous study [17]. We think that HA is formed as an intermediate phase in the synthesis but upon increasing the temperature above 1100°C it is dehydrated and undergoes a structural change, turning successively into oxyapatite and α-TCP. During heating, although the temperature of 1125°C of the polymorphic transition β-TCP→α-TCP was exceeded, this transition did not occur due to the increased thermal stability of β-TCP solid solution containing Mg2+ and Si4+. This step of phase formation could explain the fact that the unit cell parameters for the synthesized α-TCP reported above are bigger compared to ICSD # 923 (a=12.887(2)Å; b=27.280(4)Å; c=15.219(2)Å; β=126.20(1)°) instead of the expected smaller values for solid solutions containing Mg2+ and Si4+. A possible explanation is related to the uneven distribution of isomorphous impurities between the crystalline phases and, more specifically, to a negligible amount of such impurities in the intermediate HA phase, which is transformed into α-TCP in contrast to β-TCP, which contains more Mg2+ and Si4+.
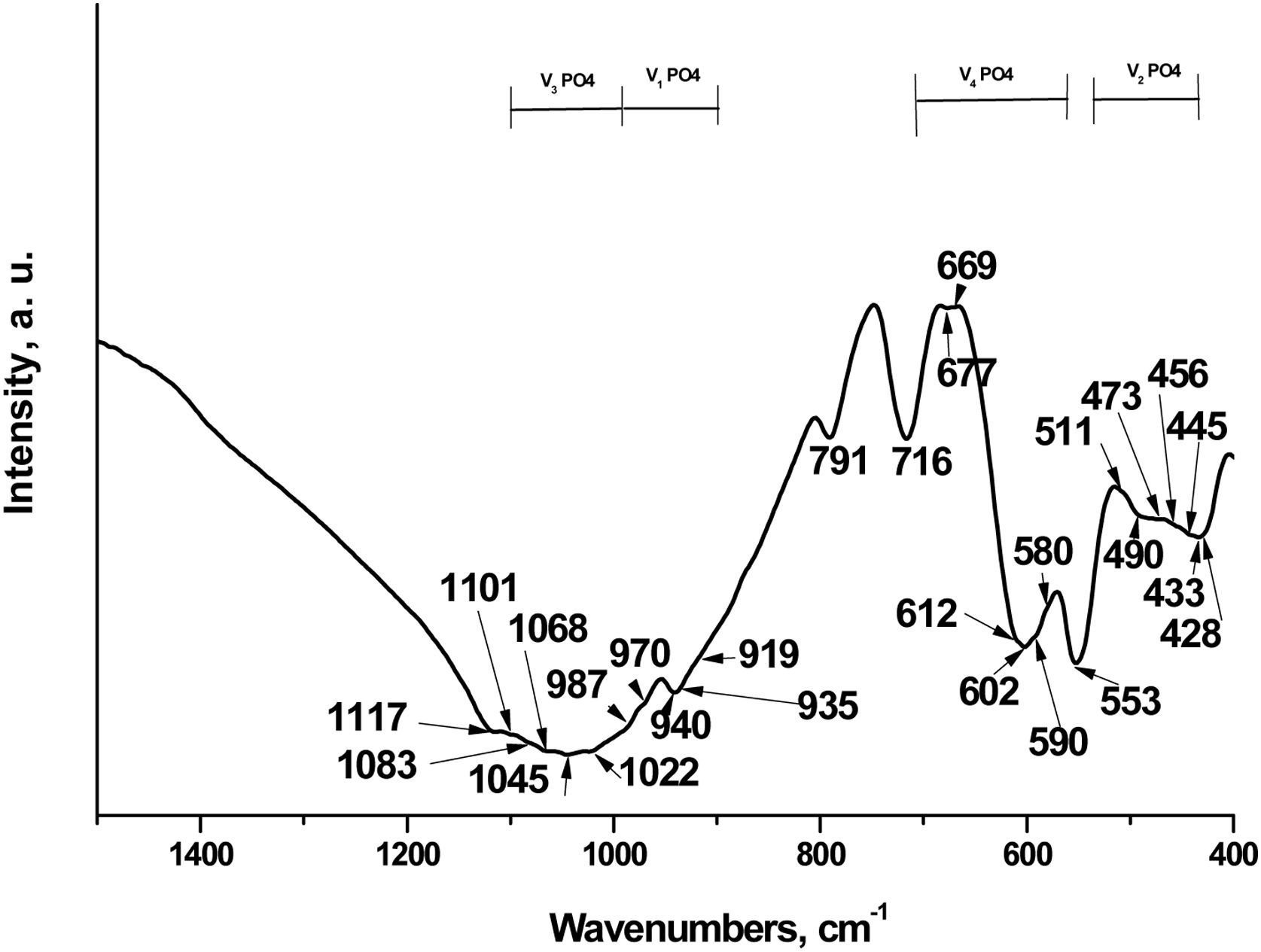
We next characterized the TCP by Fourier-transform infrared spectroscopy (FTIR). The data has been shown in Fig. 3.


According to scientific research, the FTIR spectra of the various TCPs show the presence of characteristic bands of the phosphate group, which can be attributed to three main groups of bands, respectively in the ranges 400–500cm−1, 500–700cm−1 and 900–1200cm−1. The bands at 400–500cm−1 are generally assigned to doubly and triply degenerate OPO bending mode (ν2PO43−); the bands at 500–700cm−1 are distinguished to the asymmetric PO bending mode (ν4PO43−) and those at 900–1200cm−1 to the asymmetric stretching mode from 900 to 970cm−1 (ν1PO43−) and from 970 to 1200cm−1 (ν3PO43−), in accordance with [5,11,12,24]. In addition, all ν1PO43−–ν4PO43− bands are valid from doped with different ions TCP powders. On the other hand, these bands are Raman active, in accordance with the literature data [25].
In our case, characteristic bands can be detected approximately at 428–511cm−1 (ν2PO43), 511–716cm−1 (ν4PO43−), 919–970cm−1 (ν1PO43−) and 987–1117cm−1 (ν3PO43−). These bands are also present in Raman spectra of different TCP [25,26]. The most intense band at 553cm−1 is ascribed to ν4PO43− in β-TCP [6,9]. The other intense bands, posited at 602 and 716cm−1 are characteristic of ν4PO43−, in accordance with some preliminary data. These bands are characteristic of β-TCP [5,9,24]. Furthermore, the peaks, detected at 428, 433, 445, 456, 473 and 490cm−1 (ν2PO43−), 580, 590, 612, 677cm−1 (ν4PO43−), and those at 1022, 1045, 1068cm−1 (ν3PO43−) are denoted to α-TCP in the synthesized sample [6,27]. On the other hand, the band at 716cm−1 could also be ascribed to the β- and α-TCP, in accordance with some results, published by other groups [27]. The band, posited at 791cm−1 was first identified in α-quartz [28] and was identified as evidence of the presence of an amorphous phase, which was formed after thermal treatment at 1200°C for 2h for HAP/β-TCP in accordance with our XRD results. On the other hand, Chen et al. reported that this band can be assigned to SiOSi between the two adjacent silicate tetrahedra, after immersion in SBF solution for silicon-containing glass-ceramic materials [29].
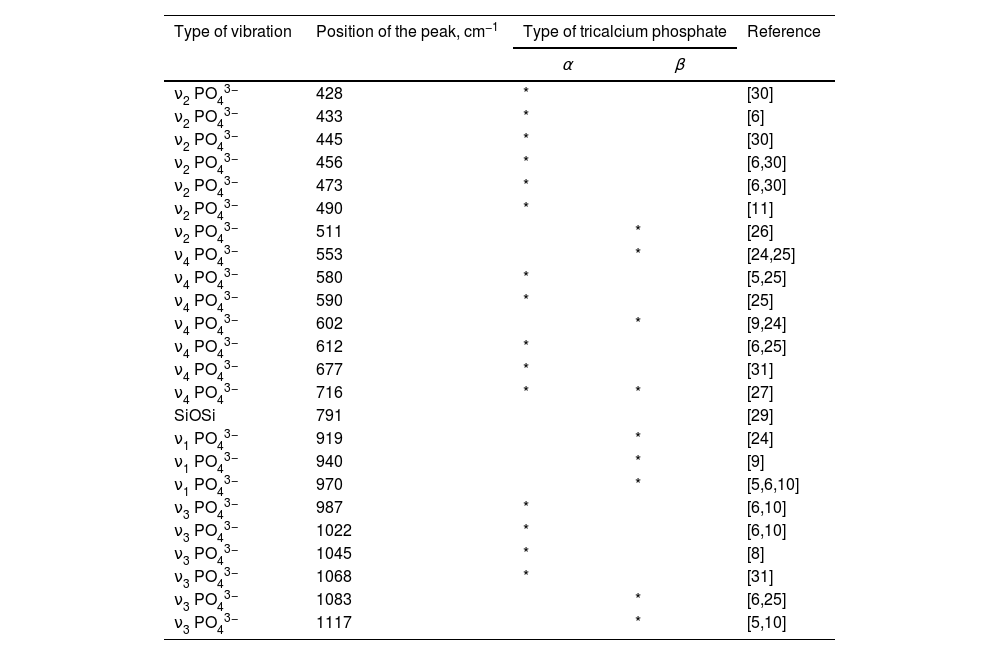
More detailed information for FTIR assignments of coexisting α/β-TCP is given in Table 1 in accordance with [5,6,8–11,24–27,29–31].
FTIR assignments for α-TCP and β-TCP for the sample, after thermal treatment at 1200°C for 2h.
| Type of vibration | Position of the peak, cm−1 | Type of tricalcium phosphate | Reference | |
|---|---|---|---|---|
| α | β | |||
| ν2 PO43− | 428 | * | [30] | |
| ν2 PO43− | 433 | * | [6] | |
| ν2 PO43− | 445 | * | [30] | |
| ν2 PO43− | 456 | * | [6,30] | |
| ν2 PO43− | 473 | * | [6,30] | |
| ν2 PO43− | 490 | * | [11] | |
| ν2 PO43− | 511 | * | [26] | |
| ν4 PO43− | 553 | * | [24,25] | |
| ν4 PO43− | 580 | * | [5,25] | |
| ν4 PO43− | 590 | * | [25] | |
| ν4 PO43− | 602 | * | [9,24] | |
| ν4 PO43− | 612 | * | [6,25] | |
| ν4 PO43− | 677 | * | [31] | |
| ν4 PO43− | 716 | * | * | [27] |
| SiOSi | 791 | [29] | ||
| ν1 PO43− | 919 | * | [24] | |
| ν1 PO43− | 940 | * | [9] | |
| ν1 PO43− | 970 | * | [5,6,10] | |
| ν3 PO43− | 987 | * | [6,10] | |
| ν3 PO43− | 1022 | * | [6,10] | |
| ν3 PO43− | 1045 | * | [8] | |
| ν3 PO43− | 1068 | * | [31] | |
| ν3 PO43− | 1083 | * | [6,25] | |
| ν3 PO43− | 1117 | * | [5,10] | |
SEM–EDX analysis of the TCP sample, after thermal treatment, is given in Fig. 4.
The SEM image demonstrated that the hollowed-out appearance of the different granules is due to occluded gaseous products during the sintering. The Ca/P ratio (at.%) observed from EDX analysis is equal to 1.48, suggesting that the thermally treated TCP sample does not contain HA. This result is in agreement with XRD and FTIR analyses.
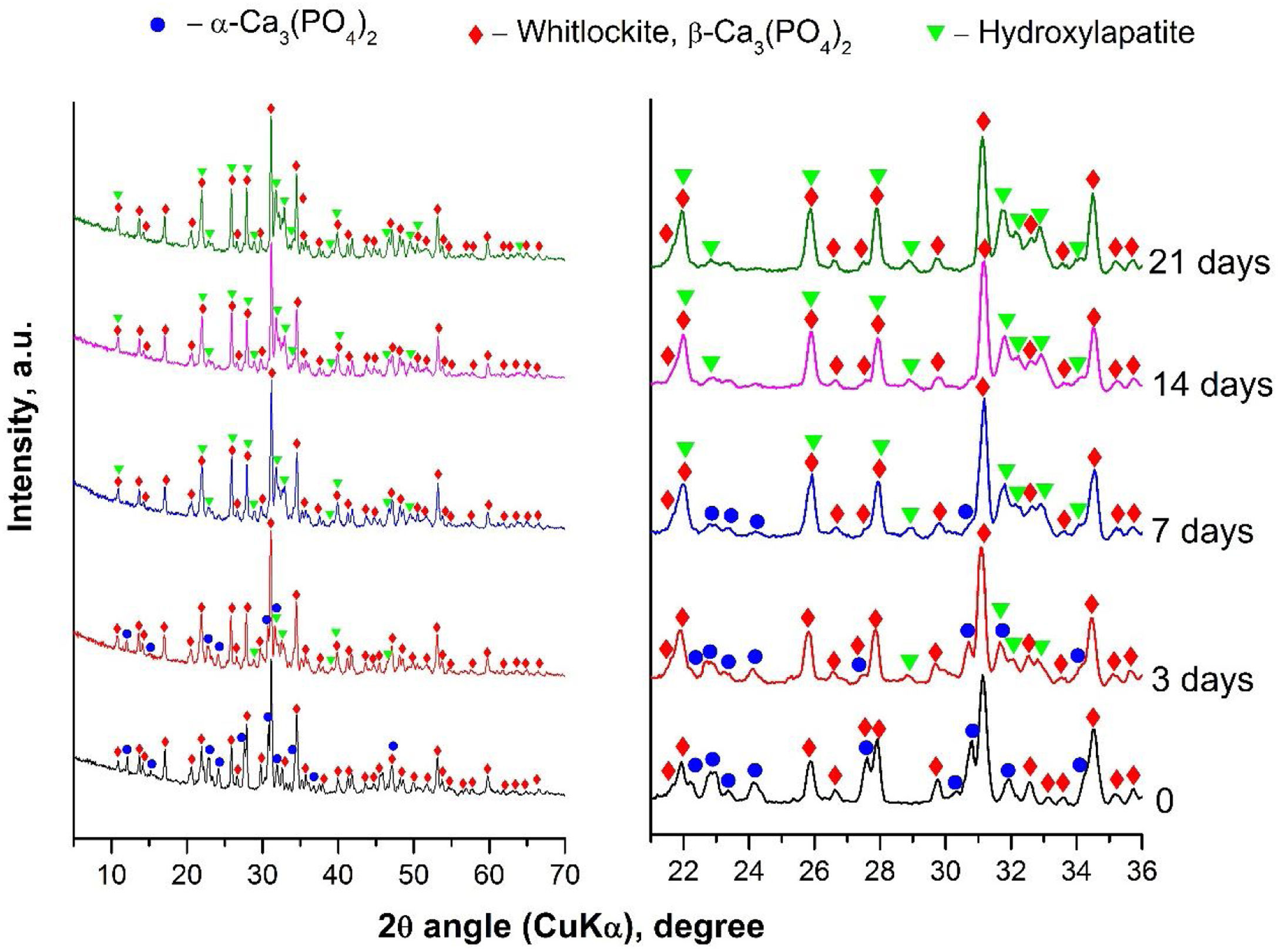
Characterization of α/β-TCP after in vitro test in SBF solutionTo evaluate the biological activity of the α/β-TCP we run XRD analyses after the immersion of the α/β-TCP in SBF solution for various time points (Fig. 5).

The XRD analyses showed that HA (PDF #01-072-1243) crystalline layer was formed on the soaked surface, after 3 days of immersion. The quantity of HA deposits increased along with the extended time of immersion, as shown in Fig. 5. However, the α-TCP phase was not observed, i.e., the α-TCP was fully dissolved in SBF solution after 7 days of soaking. Furthermore, some of the peaks for β-TCP slightly increased in intensity (for instance those at ∼22, 26 and 28 2θ) due to the overlap with the diffraction peaks of HA. Thus, the quantitative ratio between β-TCP and HA, or the sample soaked for 21 days of immersion, was 1:1. Therefore, the XRD data unequivocally confirmed the formation of HA on the surface of the synthesized by us α/β-TCP containing glass-ceramic. Likewise, the presented data are in full correspondence with the FTIR results.
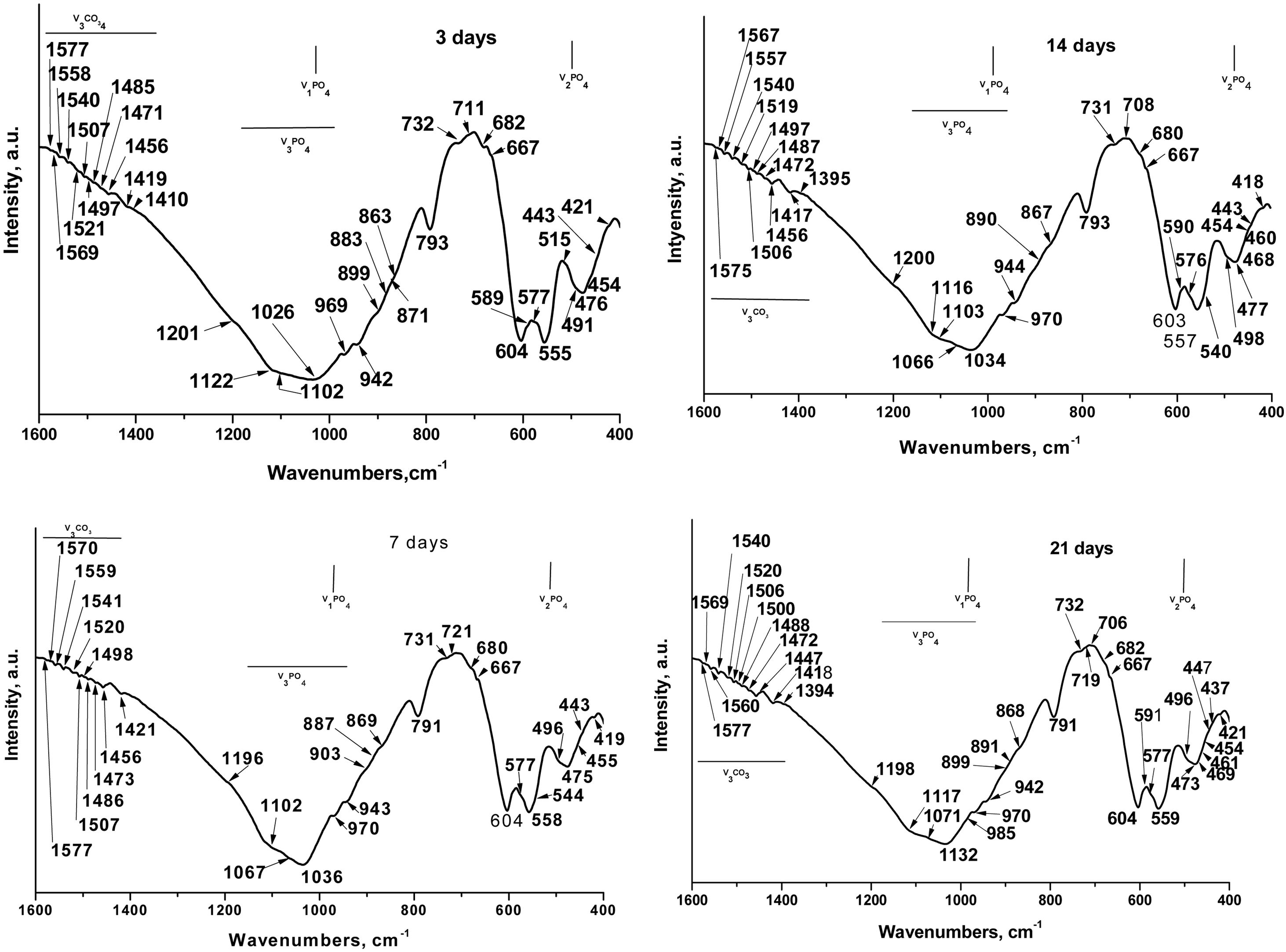
Theoretically speaking, in the FTIR spectra, for the sample soaked in SBF for various periods of time (Fig. 6) we can see that the phosphate (PO43−) exhibits four fundamental modes of vibrations: ν1PO43−, defined as the symmetric stretching mode, at ∼970cm−1[32], ν2PO43− (symmetric bending mode) at ∼470cm−1[33], ν3 antisymmetric stretching (ν3PO43−) at 900–1200cm−1[32], and ν4 antisymmetric bending (ν4 PO43−) at 540–604cm−1[34]. In addition, the carbonate (CO32−) exhibits two fundamental modes of vibrations: ν2 CO32− at ∼870cm−1[33] and ν3CO32− and ν4CO32− at ∼1390–1577cm−1[33,34].

The well-resolved ν1PO43− bands were detected at 942, 943 and 944cm−1 and at 969, 970cm−1[35]. Balamurugan et al. postulated that the band at 942cm−1 was indicative of typical HA structures [36]. Radev et al. proved that the ν1PO43− bands at 943–944cm−1 can be detected in FTIR spectra of the glass-ceramic in the CaO–SiO2–P2O5 system, after soaking in SBF solution [37]. Singh et al. related the ν1PO43− bands at 969 and 970cm−1 to the HA structures, formed on the soaked-in SBF surface of the glasses in CaO–SiO2–P2O5–Na2O–Fe2O3 system [35]. The band at 970cm−1 and those at 791 and 798cm−1 could also be related to the SiOSi stretching vibrations in nano-bio silica structures and α-quartz [29]. The band at 985cm−1 was also detected as ν1PO43− mode [32] in HA.
Nelson et al. [38] and Gasga et al. [39] reported that the ν2PO43− was doubly degenerated bending mode and two main bands were observed at ∼474 and 462cm−1. In our case, these two bands were detected at 473, 475, 476 and 477cm−1; 460, 461cm−1 and 468, 469cm−1. In addition, the other ν2PO43− were observed at 419, 421, 437, 443, 447, 457, 455, 491, 496, 498 and 515cm−1 after in vitro test in SBF solution [32,33,38]. The bands at 428, 433, 445, 490 and 511cm−1 which are indicative of the presence of α-TCP or β-TCP (see Table 1) undergoing a chemical shift to lower (from 428 to 421cm−1, and from 445 to 443cm−1) and to higher (from 473 to 475, 476 and 477cm−1, and from 490 to 491, 496, 498cm−1) wavenumbers. This “blue or red” shift caused a chemical interaction between partially dissolved TCPs and HA crystals, formed on their surface, after immersion in SBF solution. LeGeros et al. [40] prove that the ν2PO43− band at 455cm−1 are indicative of the A-type CO3HA (A-CO3HA) in which CO32− replaced OH−. The band centred at 420cm−1 can be related to the B-CO3HA in which CO32− replaced PO43−, in accordance with Ulian et al. [33].
The triply degenerated asymmetric stretching mode of the PO bond (ν3PO43−) is most sensitive to structural changes, especially in bone mineral and poorly crystalline HA. Two main bands are observed in ν3PO43− domain at 1026cm−1, 1032–1036cm−1[34,41]. In addition, the series of ν3PO43−bands at 1102, 1103, 1116, 1117, 1122, 1196, 1198, 1200 and 1201cm−1[41,42]. It is interesting to note that the CO32− band of B-CO3HA superimposes on ν3PO43− at 1071cm−1 indicates the presence of B-type carbonation, in agreement with [42]. Although, the band posited at 1002 and 1103cm−1 could also be related to the A-CO3HA, i.e. ν1CO32− band shifted slightly in all HA to 1101–1103cm−1 in the time of maturation. The obtained results indicated the presence of A-CO3HA and B-CO3HA formed during the immersion in SBF solution at the TCP surface. The bands at 1196 and 1198cm−1 can be related to the HPO42− in CDHA structure [42].
The antisymmetric bending mode (ν4PO43−) exhibits peaks in the region 560–603cm−1[43] or 546–650cm−1[44] for different CO3HA structures. It is also well known that the characteristic split bands at ∼560 and ∼605cm−1are indicative of crystalline apatites and usually prominent from well-ordered and well-defined HA crystalline phases [34,38]. In our case, the ν4PO43− bands are posited at 540, 544, 555, 557, 558, 559, 576, 577, 590, 591, 598 and 603, 604cm−1[34,41]. The band at 604cm−1 can also be ascribed to HPO42−. LeGeros related the band at 604cm−1 to the B-CO3HA [40].
In the second and third special regions modes at 800–1300cm−1 and 1350–1800cm−1 were observed in the presence of ν2CO32− at 872 and 879cm−1, and ν3CO32− at different wavenumbers from 1350 to 1800cm−1 for A- and B-CO3HA, respectively. In this context, surprising for us was the fact that the presence of the intense bands at 875–878cm−1, which are characteristic of all types of crystalline carbonate-containing apatites, was not observed. From the presented results (Fig. 6), we observed well-defined bands at 870, 872 and 873cm−1. In accordance with some FTIR results, these bands could be related to ν2CO32−[33,44]. Furthermore, some researchers concluded that the peak at 872 (873)cm−1 was indicative of A/B-CO3HA [44] and B-CO3HA [33], and the peak at 870cm−1 can be assigned to the presence of HPO42− whose ν3 PO(H) stretching mode overlapped with ν3CO32−[44]. LeGeros et al. [40] and Ou-Yang et al. [45] concluded that the band at 870cm−1 was the characteristic band for B-CO3HA of synthetic HA samples. Furthermore, we can also observe the presence of low-intensity bands at 863, 867 and 869cm−1. These bands can also be assigned to the ν2 CO32− for the samples, after in vitro immersion in SBF solution [46]. Nowiski et al., in their research for K+-containing CO3HA, reported that these bands in the region 865–872cm−1 can be associated with B-CO3HA [46]. Similar conclusions were also drawn by Aizawa et al. [47]. However, Liu et al. pointed out that the peak at 869cm−1 may be attributed to surface CO32− groups with an amorphous state, which reduced its intensity in the process of maturation [48].
The additional bands at 883, 887, 890 and 891cm−1 are also indicative of ν2CO32− modes [33]. Gangadhar et al. showed the band at 887cm−1, specific for CaCO3[49]. In our study, this band is connected only with the formation of B-CO3HA on the soaked surface because, the FTIR spectra data showed the absence of different calcium carbonates (aragonite, vaterite and/or calcite). This conclusion is in full agreement with SEM–EDX results. The absorption band at 890cm−1 could be affixed to the formation of mixed A/B-CO3HA on the immersed surface in agreement with Ulian et al. [33].
In the FTIR spectra between 1384 and 1577cm−1 the ν3CO32− are slightly visible. The values registered from us for the ν3CO32− can summarized as follows: 1384, 1385, 1386, 1390, 1395, 1417, 1418, 1419, 1421, 1447, 1456, 1471, 1472, 1473, 1485–1487, 1488, 1500, 1506, 1507, 1520, 1521, 1540, 1541, 1557, 1558, 1559, 1560, 1567, 1569, 1570, 1575, 1577cm−1 in accordance with [33,38,39,50–54].
The band at 1384cm−1 can be related either to the B-CO3HA [50]. The claim of Fereira et al., that the band at 1384cm−1 could be ascribed to the CO32− for the sample between β-TCP and bioglass [51], is very useful for our research. The peak at 1421cm−1was also related to the CO32− content in B-CO3HA [52], but Fleet et al. postulated that the small peaks at 1386 and 1390cm−1 could be related to the A/B-CO3HA [53]. Prasad et al. [54] and Montazeri et al. [55] observed that the presence of a peak at 1395cm−1 can be connected to the presence of carbonates on the immersed surface for the iron and cobalt containing 45S5/HA biocomposite [54] or the carbonate impurities on the 45S5 sample, after thermal treatment [55]. The absorption bands, positioned at 1419 and 1447cm−1 can be linked to the presence of CO32− in A-CO3HA structures [52], while the peaks at 1456, 1471, 1472 and 1473cm−1 were related to the B-CO3HA. In contrast, Paschalis et al. pointed out that the peak at 1471cm−1 can be observed in A-CO3HA [56]. On the other hand, Fleet designated that the peak at 1472cm−1 can be denoted as CO32− in A/B-CO3HA structures [53]. The peaks at 1500, 1506, 1507, 1540, 1567, and 1575cm−1 can be associated with A-type CO32− substitution in which CO32− replaced OH−[33,52] but other authors connected the peaks at 1506, 1507, 1540, 1557–1560, and 1569cm−1 with B-type CO32− substitution in which CO32− replaced PO43− in B-CO3HA lattice [40,53]. In addition, Ulian et al. underlined that the peaks in the region 1557–1560cm−1 can be referred to as the CO32− modes in mixed A/B-CO3HA [33]. Altogether the studies of other groups performed a back bound of our analyses. Based on our data (Fig. 6), we can conclude that:
- •
A/B – type carbonate containing CDHA (A/B-CO3CDHA) was formed on the soaked surface.
- •
No other CaCO3 phases, such as vaterite, aragonite, or calcite, were present when A/B-CO3CDHA was formed on the surface.
- •
The bands at 428, 433, 445, 490 and 511cm−1 which are indicative of α- and β-TCP undergo a “red” or “blue” shift, caused by the chemical interaction between partially dissolved β-TCP and fully dissolved α-TCP.
- •
The band at 716cm−1 could be ascribed to the α-TCP as this band fully disappeared after immersion in the SBF solution.
The obtained FTIR results corresponded to the XRD and SEM data.
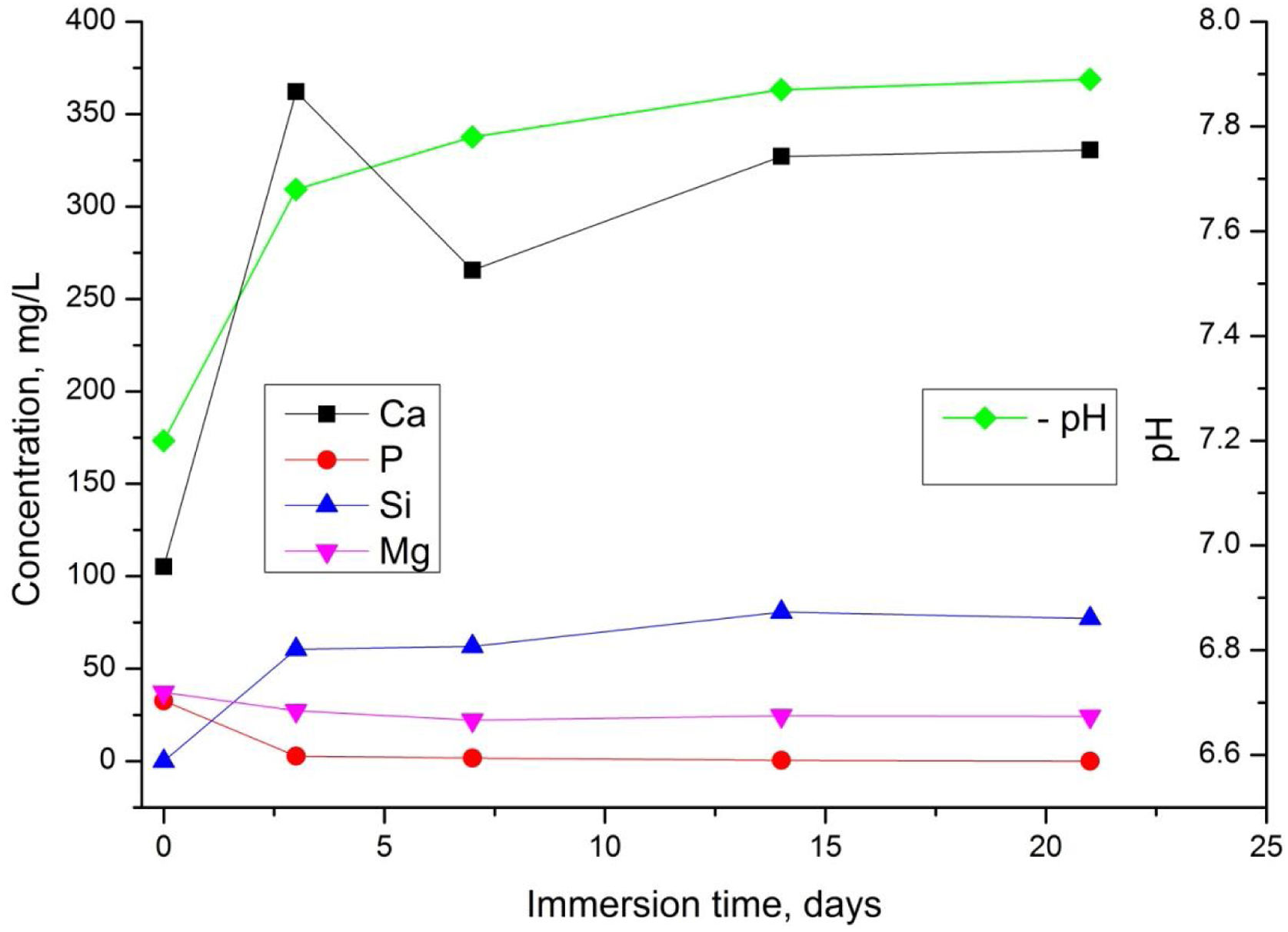
We next investigated the behaviour of the TCP sample dissolution in SBF (Fig. 7).

It is well-known that the dissolution rate of different biomaterials depends on multiple aspects including microstructure, crystal phases and pH. From Fig. 7, the values in the SBF solution exhibited a slight increase from about 7.2 to 7.8 until 21 days. The variation of Ca2+and Si4+ ions increased rapidly during 3 days, then the quantity of Ca2+decreased slightly probably due to the suppressive effect of Mg2+ and Si4+ which are presented in the sample [5].
On the other hand, the formation of carbonated hydroxyapatite is in direct relation to the released Ca2+ and Si4+ ions in the SBF solution. However, these ions are the main factor for the rapid bonding of the ceramic to the damaged tissue surface [57]. Moreover, it is noticeable that the concentration of Mg2+ in the SBF solution decreases, which can be related to the already established fact that Mg2+ decelerate the dissolution process of magnesium-containing ceramics, while Na+ and K+ facilitate/favour the hydrolysis process [58]. After 7 days of soaking the concentration of Ca2+and Si4+remained almost unchanged. The quantity of P5+ slightly decreased up to day 3 and remained almost constant thereafter.
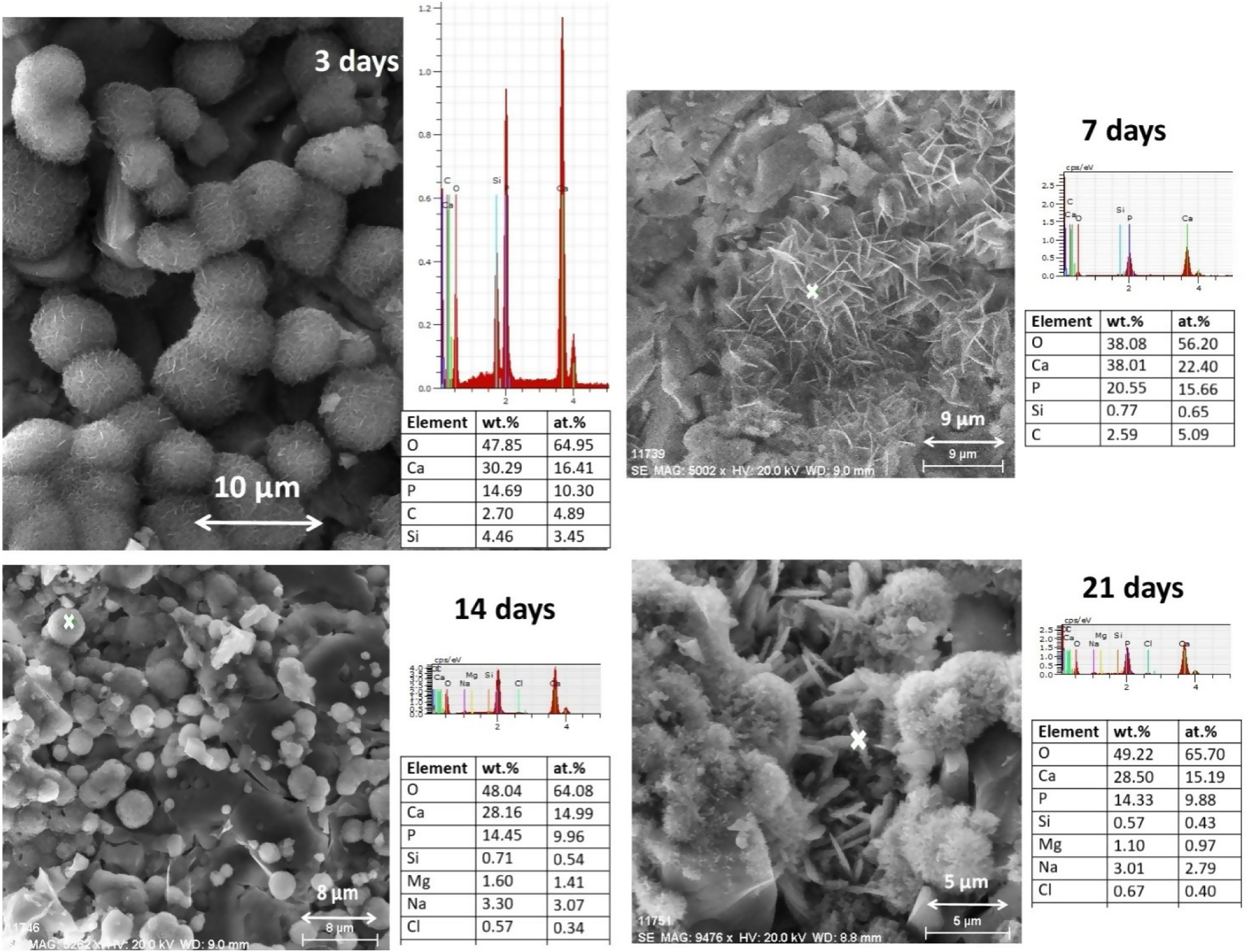
One of the significant characteristics of biomaterials for bone regeneration is their ability to bind with living bone by forming apatite on their surface [4,5]. The formation of HA on the α/β-TCP bioceramic after the SBF solution immersion under static conditions was evaluated using SEM and EDX (Figs. 8 and 9).
 after SBF immersion for various time points in static conditions.")
 after SBF immersion.")
As shown in Fig. 8, the synthesized coexisting α/β-TCP specimens after 3 days of soaking displayed the typical microstructure under SEM. The structure of the HA layer was similar in composition and structure to CDHA. It is observed that the crystals were poorly crystalline [5]. Aggregates with a spherical morphology were found which are composed of very small crystals. As can be seen from the SEM images in Fig. 8, the morphology of the CO3HA surface layer is different. It could be directly related to the content of CO32− ions in it. As noted by R.Z. LeGeros et al., the increasing content of CO32− ions in the apatite layer results in a change in the morphology of the sample, namely: at a lower content of CO32− the spherical morphology is dominant. Obviously, our result is in agreement with the conclusion of the authors [59]. From EDX results (Fig. 9) it is visible that the Ca/P ratio (at. %) is equal to 1.59 indicating the CDHA was formed on the surface. From the SEM image for 7 days of soaking, the plate large HA crystal clusters with flower-like structures were formed. Likewise in the SEM image, the HA particles became interconnected on the surface of the microporous sample. The Ca/P ratio was 1.50 confirming that he CDHA was formed. After 14 days of immersion SEM depicted the presence of a well-defined structure of the immersed sample with Ca/P ratio equal to 1.56, proposing that the CDHA was formed persistently and at day 21 of soaking, the well-defined CDHA particles were formed along with consistent Ca/P ratio 1.54. Although in some of the analyses presented in Fig. 9 Mg was not registered, the samples do not differ by this indicator. Probably the reason for not being detected at some points is related to the fact that the Mg content of the samples is close to the detection threshold of the applied EDX-analysis.
From the obtained SEM–EDX results we concluded that the CDHA were formed on the soaked surface of the sample and these data confirmed the XRD and FTIR analyses.
Effect of α/β-TCP on cell viability of BM cells and pre-osteoclastsThe XRD, FTIR and SEM–EDX analyses of the coexisting α/β-TCP showed the presence of less than 1wt% Mg2+. After immersion in the SBF solution, which mimics the human plasma, HA was formed on the surface of the α/β-TCP. During this process Ca2+ and Si4+ ions increased in the solution within 3 days of the immersion. The presence of some Mg2+ and Ca2+ and Si4+ ions, the characteristics of the α/β-TCP and the formation of the surface HA, all are key factors determining the TCP bioactivity and toxicity, and its implication in tissue engineering for bone regeneration and repair. Therefore, we investigated the cytopathic effect of the TCP on murine bone marrow cells and pre-osteoclasts (Fig. 10A, B).
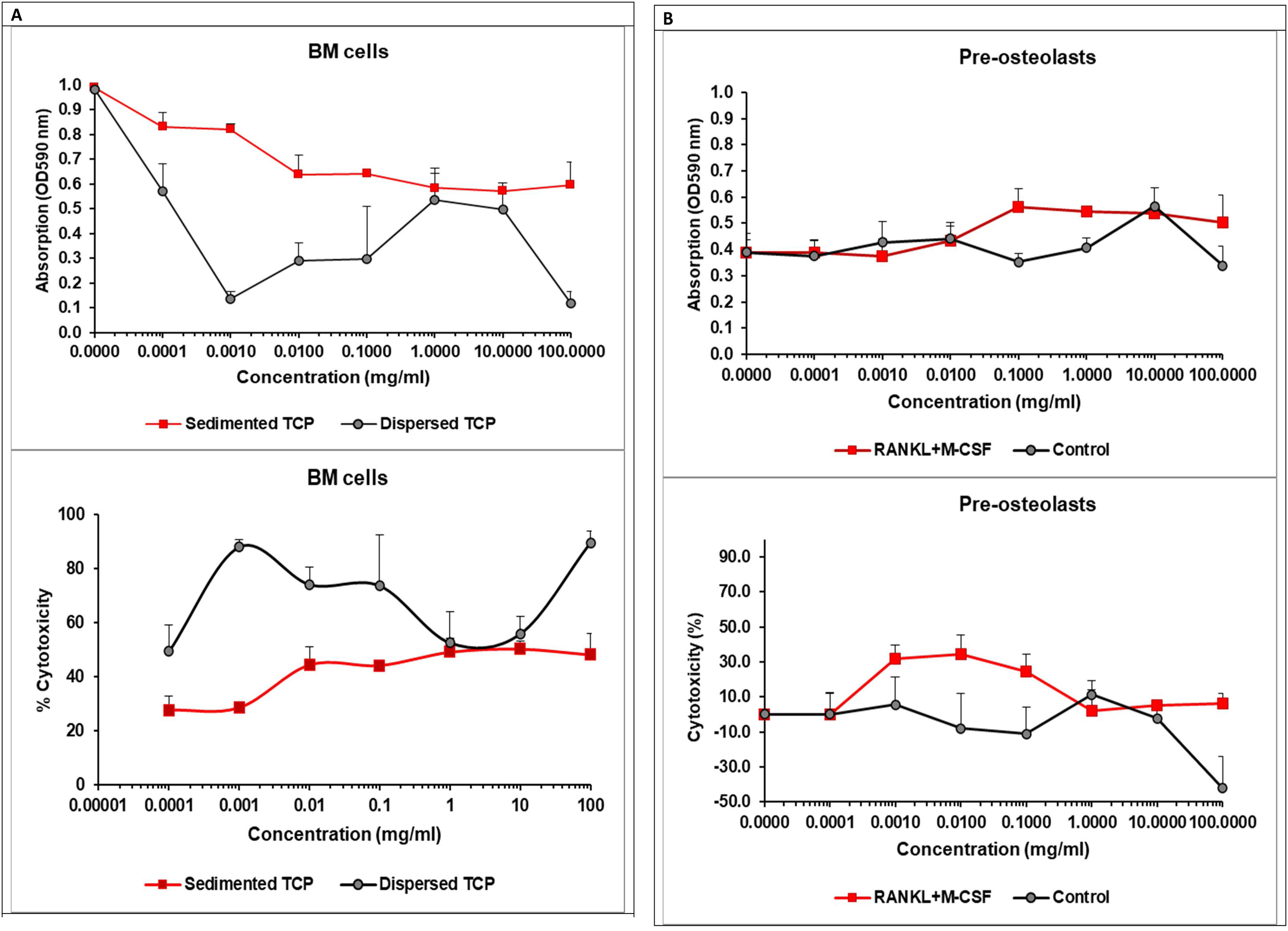
 Freshly isolated murine BM cells were seeded at a concentration of 5×104/ml in 96-well plate in the presence of increasing concentration of the TCP. Cell viability was assessed after 40h of incubation by colorimetric assay using MTT kit. Data represent mean±SD of a sample run in 4 repeats. The experiment was performed three times (n=12/sample). (B) Pre-osteoclasts were generated from BM cells upon culturing in the presence of M-CSF, followed by supplementation or not with 50ng/ml RANKL and 30ng/ml M-CSF (to drive or not the osteoclast differentiation). α/β-TCP was added to the plate at increasing concentrations 30min before the cell suspension. After 48h, the cell viability was assessed as described in (A). Data represent mean±SD of a sample run in 4 repeats. The experiment was performed three times (n=12/sample).")
The effect of the α/βTCP on cell cytotoxicity. (A) Freshly isolated murine BM cells were seeded at a concentration of 5×104/ml in 96-well plate in the presence of increasing concentration of the TCP. Cell viability was assessed after 40h of incubation by colorimetric assay using MTT kit. Data represent mean±SD of a sample run in 4 repeats. The experiment was performed three times (n=12/sample). (B) Pre-osteoclasts were generated from BM cells upon culturing in the presence of M-CSF, followed by supplementation or not with 50ng/ml RANKL and 30ng/ml M-CSF (to drive or not the osteoclast differentiation). α/β-TCP was added to the plate at increasing concentrations 30min before the cell suspension. After 48h, the cell viability was assessed as described in (A). Data represent mean±SD of a sample run in 4 repeats. The experiment was performed three times (n=12/sample).
Pre-osteoclasts were differentiated from BM cells in the presence of 30ng/ml M-CSF for 48h. We evaluated cell viability of BM cells or pre-osteoclasts in the presence of increasing concentrations of α/β-TCP ranging from 0.0001 to 100mg/ml using a colorimetric assay (MTT assay) estimating cellular metabolic state. After 40h (for BM cells) or 48h (for pre-osteoclasts) of incubation, the cell viability was assessed and the percentage of cytotoxicity was calculated as described in Material and Methods section (Fig. 10A).
The MTT assay demonstrated that α/β-TCP decreased cell viability of BM cells in a dose-dependent manner with the lowest cell survival occurring at 100mg/ml. The cell cytotoxicity induced by the highest concentration of α/β-TCP, however, was less than 50% (Fig. 10A). BM cell suspension is heterogenous, consistent of various hematopoietic progenitors, mature immune cells, mesenchymal stem cells, reticulocytes and stromal cells, all forming specific bone marrow niches. Considering this heterogeneity, we think that α/β-TCP had a distinct impact on viability of various populations. α/β-TCP may reduce the viability of hematopoietic progenitor cells, which strongly depend on various growth factors for cell survival and commitment, thus being more sensitive. Hypothetically, if α/β-TCP affects the viability of hematopoietic progenitors, this might be beneficial for bone repair, as less mature inflammatory cells might be generated when the material is applied in vivo. By opposite, mesenchymal stem cells (MSCs) may have accelerated cell survival, up to 120% in the presence of β-TCP at day 3 of culture as shown by Choy at al. [60]. TCP can induce changes in the MSCs morphology too by providing a surface for attachment of newly generated osteoblast progenitors [60]. It is intriguing that TCP with high microporosity can inhibit osteoblast differentiation while non-microporous ceramics cannot [61]. Thus, we speculated that our α/β-TCP, showing improve density properties, might be a suitable material for stimulation of osteoblastogenesis. However additional investigations are needed to confirm this statement.
While MSCs and osteoblasts might be clearly related to the beneficial effect of TCP on bone regeneration, osteoclasts might compromise the impact of TCP on bone. Thus, we evaluated the survival of pre-osteoclasts in the presence of increasing concentrations of TCP (Fig. 10B). RANKL and M-CSF are growth factors sustaining osteoclast viability independently of the presence of TCP. We observed that TCP at low concentrations increased cytotoxicity of pre-osteoclasts cultured in the presence of RANKL and M-CSF suggesting that TCP may impact early osteoclast differentiation. When pre-osteoclasts were cultured in the absence of the growth factors, RANKL and M-CSF, the TCP increased cell survival in a dose-dependent manner probably affecting the viability of monocyte precursors but not their differentiation to osteoclasts (Fig. 10B).
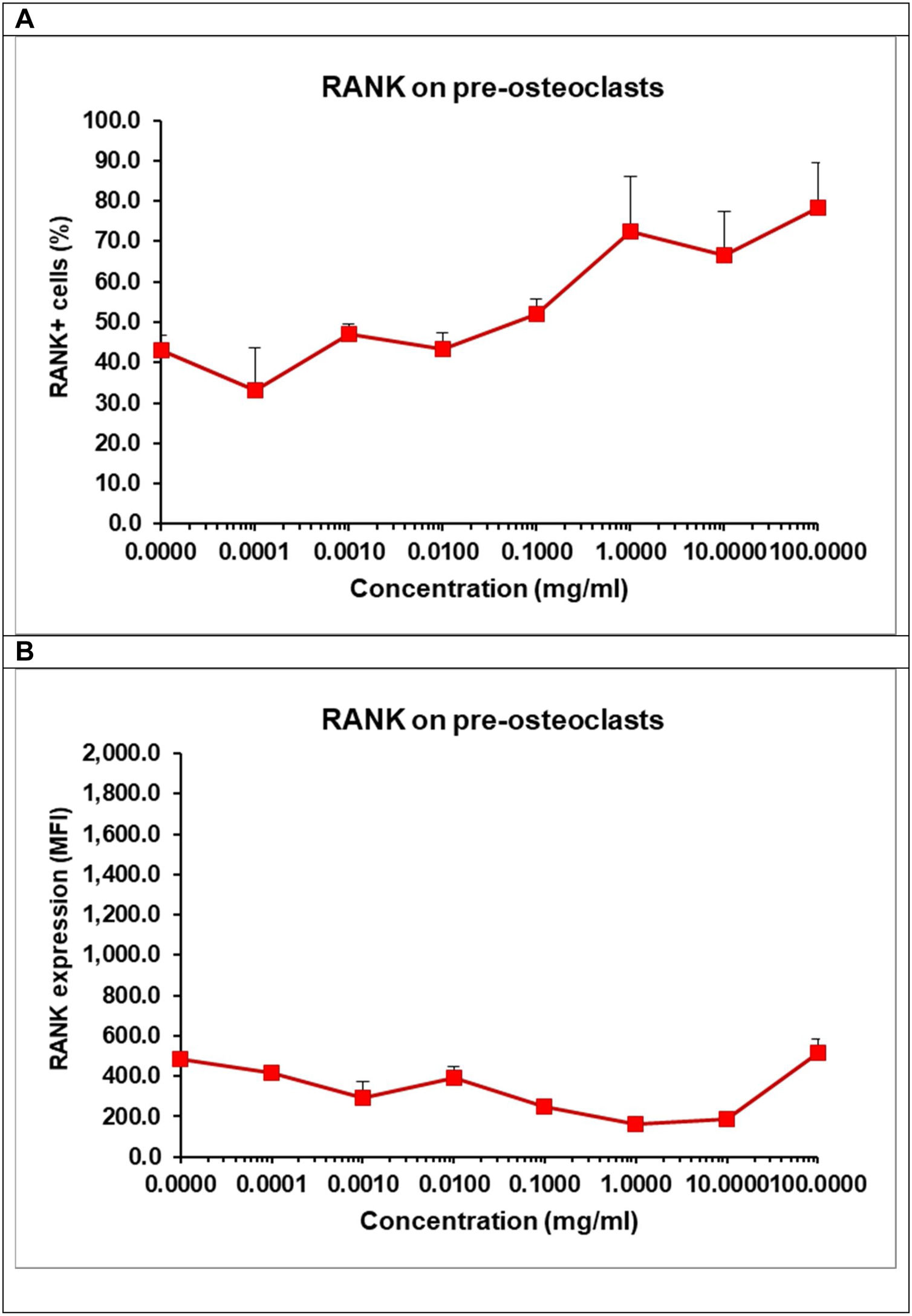
Effect of α/β-TCP on the expression of RANK and TRAIL on pre-osteoclastsIn order to understand whether TCP affect pre-osteoclast commitment and differentiation, we evaluated the intensity of the surface expression of the receptor RANK on osteoclasts as well the percentage of RANK+ cells by flow cytometry (Fig. 11A, B). We cultured the pre-osteoclasts in the presence of growth factors RANKL and M-CSF and in the presence of TCP at various concentrations for 48h. The surface expression of RANK on pre-osteoclasts remained low and almost unchanged as RANK might be constantly engaged by RANKL present in the media for 48h (Fig. 11A). Osteoclasts need a constant signal delivered via RANK-RANKL interaction to become mature osteoclasts and our data showed that the presence of TCP failed to compromise directly this process. Interestingly, we observed that TCP increased in a dose-dependent manner the percentage of RANK+ cells indicating that TCP stimulates early RANKL-induced osteoclast differentiation by elevating the number of osteoclast-like cells (Fig. 11B).
 and the surface expression of RANKL (B). Pre-osteoclasts were incubated with 50ng/ml RANKL and 30ng/ml M-CSF in the presence of increasing concentrations of TCP. After 48h, the frequency of RANK+ cells and the surface expression of RANK were determined by flow cytometry. Data represent mean±SD of a sample run in triplicates. The experiment was performed three times (n=9/sample).")
Effect of TCP on the frequency of RANK+ cells (A) and the surface expression of RANKL (B). Pre-osteoclasts were incubated with 50ng/ml RANKL and 30ng/ml M-CSF in the presence of increasing concentrations of TCP. After 48h, the frequency of RANK+ cells and the surface expression of RANK were determined by flow cytometry. Data represent mean±SD of a sample run in triplicates. The experiment was performed three times (n=9/sample).
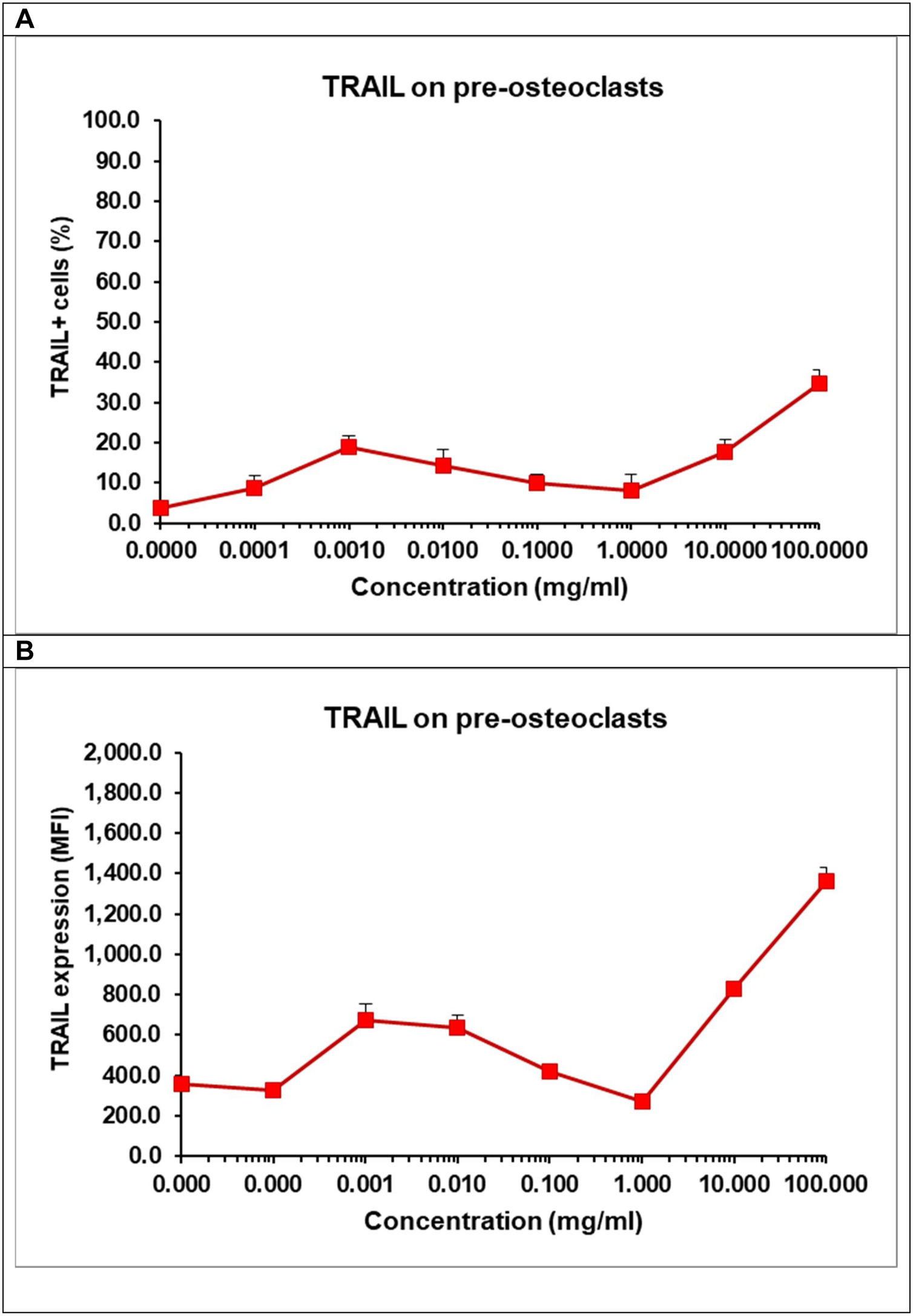
Differentiation of osteoclasts can be modulated by RANK, RANKL and TRAIL. The latter is a receptor from the tumour necrosis factor (TNF) family with a death domain triggering apoptotic signals. TRAIL is an important factor for the survival of osteoclasts [62]. We observed that at low concentration TCP decreased cell viability of pre-osteoclasts (Fig. 10B), but it also increased the percentage of RANK+ osteoclasts. As these results seems controversial, we evaluated the effect of TCP on the number of TRAIL+ cells (Fig. 12A) and the surface expression of TRAIL on pre-osteoclasts (Fig. 12B).
 and the surface expression of TRAIL (B). Pre-osteoclasts were incubated with 50ng/ml RANKL and 30ng/ml M-CSF in the presence of increasing concentrations of TCP. After 48h, the frequency of TRAIL+ cells and the surface expression of TRAIL were obtained by flow cytometry. Data represent mean±SD of a sample run in triplicates. The experiment was performed three times (n=9/sample).")
Effect of TCP on the frequency of TRAIL+ cells (A) and the surface expression of TRAIL (B). Pre-osteoclasts were incubated with 50ng/ml RANKL and 30ng/ml M-CSF in the presence of increasing concentrations of TCP. After 48h, the frequency of TRAIL+ cells and the surface expression of TRAIL were obtained by flow cytometry. Data represent mean±SD of a sample run in triplicates. The experiment was performed three times (n=9/sample).
Our data showed that, TCP, at higher concentrations – 10–100mg/ml increased almost 3 times the percentages of TRAIL+ osteoclasts (Fig. 12A) and the surface expression of TRAIL (Fig. 12B) confirming that TCP may affect cell viability of pre-osteoclasts (Fig. 10B). We think that TCP may accelerate the generation of RANK+ cells, but it also can induce an upregulation of TRAIL that might trigger a backup mechanism of a TRAIL-dependent regulation of the cell survival during differentiation [62]. TRAIL can be expressed at cell surface but it can be also secreted and released at late stage of osteclastogenesis, in turn, triggering apoptosis of osteoclasts at late stage [62].
The effect of TCP on osteoclasts should not be considering as a reason TCP to be less effective to support osteoblastogenesis. Indeed, our data corresponded to the study evaluating the effect of calcium phosphates on osteoclast formation and on the osteoclast-osteoblast crosstalk [63]. Similarly, to our data showing that TCP increased the percentage of RANK+ osteoclasts, the authors demonstrated that β-TCP stimulated the generation of multinucleated osteoclasts [63]. They also found that during osteoclast culture, the distinct chemical environment induced by the calcium phosphate phases affected the crosstalk between osteoclasts and osteoblasts [61].
Important factors which can determine the TCP effect on cell viability and osteoclast differentiation in our study is the release of Ca2+, Mg2+ and Si4+. It has been shown that Mg2+ improves the effect of TCP on osteoblasts, the performance of the scaffolds for bone regeneration processes, angiogenesis and osteogenesis [64]. The osteoclasts resorbed slowly the TCP doped with high amount of Mg2+ thus many attempts have been made to improve the β-TCP with doped Mg2+[65]. In a study using human but not murine osteoclasts, the β-TCP composite with Mg(OH)2 and poly(lactide-co-glycolide) (PLGA) inhibited the differentiation of pre-osteoclasts into osteoclasts. Our explanation for this discrepancy is the fact that the composite in this study and our α/β-TCP material contained various amounts of Mg2+ (6% vs 1%, respectively). On the other hand, the studied materials contain different phases: β-TCP in [64] and α-TCP, β-TCP crystalline phases in a vitreous silicate matrix in our case.
The immersion of our α/β-TCP in cell culture media may result in a rapid increase of Ca2+ (similarly to the SBF solution) within 2–3 days, time points selected for evaluation of RANK+ cells by flow cytometry. It has been shown that the elevated extracellular Ca2+ increased the intracellular Ca2+ levels in osteoclasts leading to alteration in intracellular signalling and decreasing resorptive potential of mature osteoclasts [66]. In our study, pre-osteoclasts were incubated with TCP for 48h, a time point when Ca2+ was still increasing. Therefore, we cannot exclude that Ca2+ would fail to impact the terminal differentiation of osteoclasts obtained for 7 days of culturing. Similarly, the presence of Si4+ may diminish cell growth of osteoclasts in long term cultures [67]. Our study showed that the total BM cell population was more sensitive to TCP than pre-osteoclasts. The survival of pre-osteoclasts at initial differentiation for 48h with RANKL and M-CSF was modestly abrogated by TCP. We found an increased frequency of RANK+ cells by TCP indicative of accelerated RANKL-mediated initial osteoclast differentiation. At higher concentrations, TCP increased the expression of TRAIL. It can trigger apoptosis of osteoclasts when released in the medium. Thus, we think that TCP at high concentrations may trigger a mechanism for regulation of osteoclast apoptosis/survival during the osteoclast differentiation.
We found that the TCP α/β form may affect differently the apoptosis and early differentiation of osteoclasts. However, β-TCP can stimulate the generation of multinucleated osteoclasts while α-TCP was mainly related to osteoblast activation [63]. Indeed, an effective delivery system of encapsulated calcium phosphate nanoparticles increased a potent osteogenic response in osteoblasts [68]. Despite we observed solely the effect of α/β TCP on pre-osteoclasts, we hypothesized that the material might be used for implantation. Bone homeostasis and regeneration depends on the tight balance between osteoclasts and osteoclasts rather than on the stimulation and presence of the single bone cell type. The survival, activation and differentiation of osteolasts/osteoblasts are reciprocally regulated via soluble factors [69] and direct spatiotemporal intercellular interactions [70]. The imbalance in those three processes might damper the bone homeostasis with in excess in degradation or bone formation. Thus, the materials used for implantation might not only affect regeneration by inhibiting osteoclasts by also they have to prevent the excessive bone formation along with balanced osteoclastogenesis.
ConclusionsA monolithic and sintered glass-ceramic (α/β-TCP) was obtained, which contained two polymorphic modifications of tricalcium phosphate: α-TCP and β-TCP as main crystalline phases and silicate glass. The EDS results proved that magnesium and silicon were included as isomorphous impurities in the TCP structure. The conducted experimental research confirmed the role of silicon dioxide and magnesium oxide on the polymorphic transformation temperature, sintering process and densification of ceramics. By means of XRD, FTIR, SEM and EDS, the ability of glass-ceramic to form a hydroxyapatite layer on its surface during an in vitro bioactivity test in SBF was proven. The HA layer formed was similar in composition and structure to carbonate containing calcium-deficient hydroxyapatite. The ICP-OES results showed the high solubility of glass-ceramic in SBF. The higher solubility of α-TCP and the stability of β-TCP by the end of the 21-day test period in SBF were established by XRD and FTIR. TCP showed a dose-dependent cytotoxic effect on murine BM cells and modest impact on the viability of pre-osteoclasts cultured in osteoclast differentiation media. TCP increased the percentage of RANK+ osteoclasts. It elevated the expression of TRAIL and the number of TRAIL+ cells. In conclusion, the TCP showed the potential to affect the survival/differentiation of pre-osteoclasts at early stage of osteoclastogenesis a key process for balanced regulation of bone remodelling and repair. Despite that our data show that the new glass-ceramic material might be used in tissue engineering as coating material or scaffold, further studies should be conducted to investigate its effect on bone homeostasis and cross-talk between osteoclasts/osteoblasts.
This study is funded by the European Union-Next Generation EU, through the National Recovery and Resilience Plan of the Republic of Bulgaria, project № BG-RRP-2.004-0002, “BiOrgaMCT”.

