




Mineral carbonation of construction and demolition waste is a viable alternative for the reduction of CO2 emissions from industry. Concrete and cement, together with ceramic blocks, are the primary sources of calcium for mineral carbonation in which CO2 is fixed in a stable and inert process. One type of concrete was selected from 5 for the carbonation tests. Crushed, separated into size fractions, moistened to 20% and tested at 10 bars of CO2 for 24 to 720h. Destruction of portlandite and ettringite phases was determined. Calcite precipitated as carbonate phase. Maximum carbonation was reached after 72h, fixing 6.5% of CO2.
La carbonatación mineral de los residuos de construcción y demolición es una alternativa viable para la reducción de las emisiones de CO2 de la industria. El hormigón y el cemento, junto con los bloques de cerámica, son las principales fuentes de calcio para la carbonatación mineral en la que el CO2 se fija en un proceso estable e inerte. Se seleccionó un tipo de hormigón de entre 5 para las pruebas de carbonatación. Triturado, separado en fracciones de tamaño, humedecido al 20% y probado a 10 bares de CO2 durante 24 a 720 horas. Se determinó la destrucción de las fases de portlanita y ettringita. La calcita se precipitó como fase de carbonato. La máxima carbonatación se alcanzó después de 72 horas, fijando el 6,5% de CO2.
Climate change is a long-term challenge, but it demands an urgent action due to the growth of greenhouse gas emissions to the atmosphere and its accumulation rate, increasing the global average temperature. This kind of emissions to the atmosphere and the risk on an increase in global average temperature are mainly since carbon dioxide concentrations have increased by 40% since the pre-industrial period due to anthropogenic effects [1,2]. To avoid or mitigate these figures, CO2 emissions should be minimized. The main measures proposed to lead to a reduction in the energy consumption of fossil fuels by offsetting emissions from industrial processes, promoting the use of energy from non-polluting sources and encouraging the use of technologies for carbon capture, utilization and storage (CCUS) [3]. These strategies imply the development of innovative, available and profitable techniques.
Construction and demolition waste (C&D waste) mainly come from building demolitions, construction materials rejected for new plant works, as well as non-reusable or defective products generated during manufacturing processes [4,5]. C&D waste is a potential CO2 material to capture CO2. Thus, previous researches [6–9] have analyzed their ability to react with CO2 by mineral carbonation of calcium and/or magnesium silicates, oxides and hydroxides, present in their composition.
Mineral carbonation offers a stable and safe alternative to the use of underground geological formations for the storage of this greenhouse gas. This method of CO2 capture and storage has been considered for years as a topic goal in cement chemical investigations [10]. It is a complex physicochemical process that induces a slow modification of the concrete structure and, over time, develops changes in its physical [11,12] and chemical [13–18] properties. The reactions that occur in the concrete achieve the complete stabilization of CO2, by chemical fixation of the gas as new minerals phases:
CO2 remains in a solid-state through the formation of carbonates such as calcite, magnesite or dolomite. These minerals are very abundant in the earth crust, and they are stable on a geological timescale of millions of years. The carbonation of minerals which contain calcium and magnesium is a natural spontaneous reaction, although it occurs on geological timescales [19]. The challenge is to accelerate these natural reactions to the point of fixing CO2 at the same rate that it is generated in the consumption of fossil fuels.
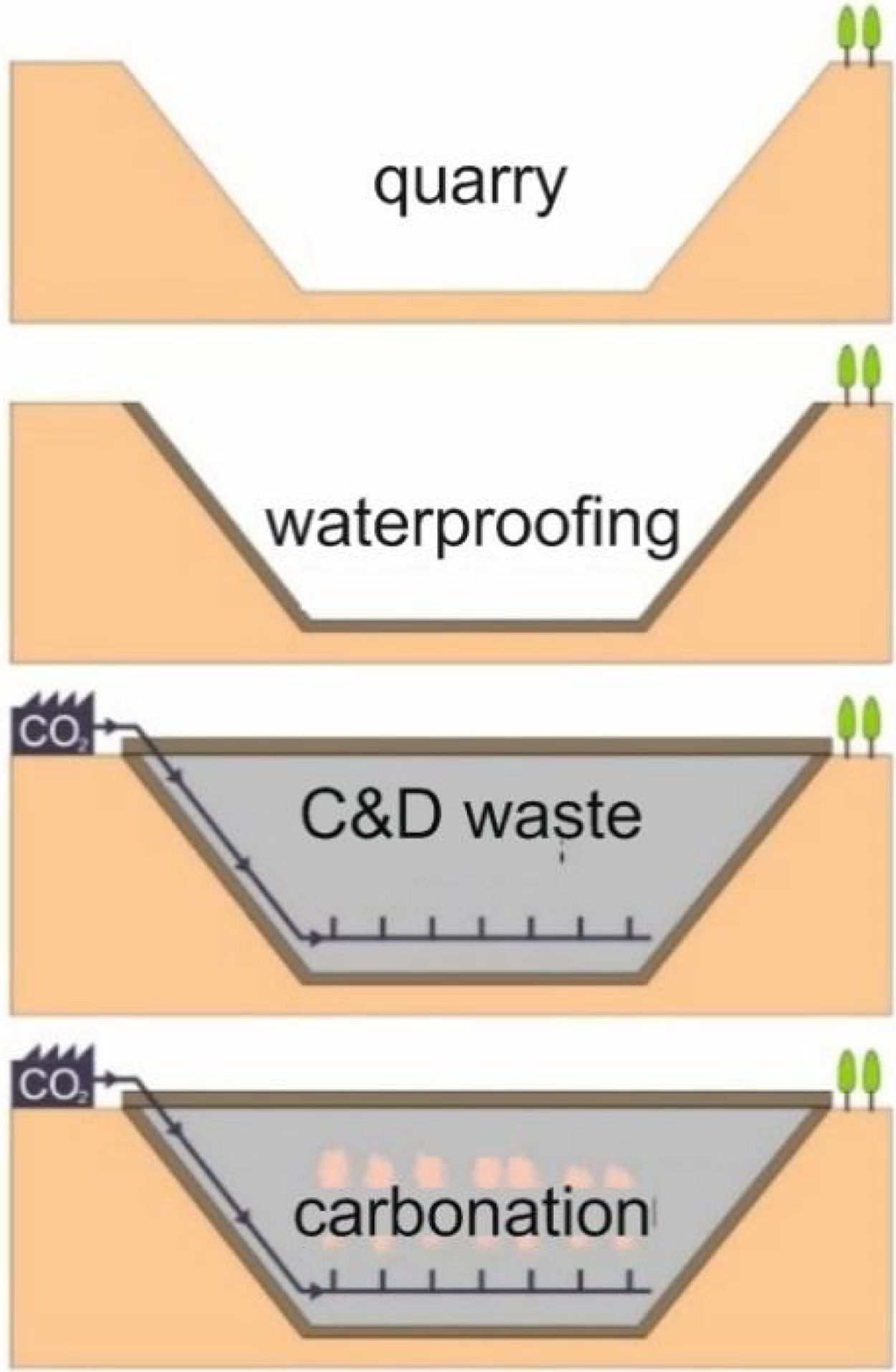
This research presents considerations about the capture of CO2 in conventional concrete buildings through a laboratory simulation based on researches carried out by Martín et al. [4,8,20,21]. These works reproduce the conditions that should occur in a waterproofed quarry vessel to optimize the mineralogical carbonation of the C&D waste. Previous to the filling phase of the quarry vessel [4], all the quarry hole should be waterproofed with clay materials before dumping C&D carbonate waste material according to the proposed model. Within the landfill it would be established a system of transport, distribution and diffusion of CO2; finally, to avoid the gas lost, the upper part of the vessel must be waterproofed with the same material. The gaseous CO2 may be brought into contact with the C&D waste and the carbonation reaction would occur (Fig. 1).

In this work, the possible mechanisms of CO2 capture in concrete, which together with ceramic blocks or bricks, are the main components of C&D waste, have been studied. Tests have been evaluated at room temperature and low pressures, depending on the type of concrete and particle size, reproducing a process that can potentially be economical and profitable on a large-scale and environmentally suitable. Results of this research contribute to the application of carbonation of C&D waste on an industrial scale, particularly if that wastes are used as material to fill natural deposits, as depleted quarries. As it was noted above, CO2 could be injected in quarries and develop carbonation reactions.
Materials and methodsConsidering that the concrete carbonate phases come from cement, concretes with aggregates of different nature (siliceous and limestone) were studied, to assess the behaviour and carbonation possibilities in both cases. The main characteristic property of concrete is the compressive strength at 28 days (UNE-EN 12390-3:2003) [22], whose value expressed in MPa is used to name this type of materials.
Thus, following the incidence of cement in the compressive resistance, concretes of different strengths, within the most used ranges for building, have been considered for the study. Materials used for the study are shown in Table 1.
To facilitate the treatment of the materials, concrete samples were crushed in a jaw crusher and later homogenized. After being quartered, they were classified into three groups by their maximum particle size (2mm, 1mm and 0.5mm). For its chemical and mineralogical characterization, the grinding of the samples was carried out in agate mortar and the resultant material was sieved to 50μm.
The carbonation tests as gas-liquid-solid phase reaction, were carried out in a 0.3L volume hermetic reactor (Parr Instrument Co., Moline, IL, USA). The fixed conditions were 10 bars of CO2, a 4:1 solid-water ratio (20% humidity) and room temperature. The variable conditions were the reaction time (between 24 – 1 day – and 720h – 30 days –), and the particle size.
Material characterization and carbonation process tracingThe major elemental composition expressed in oxides was performed by X-ray fluorescence (XRF) with an automated Panalytical Axios (Marverl Panalytical Ltd, Netherlands) model spectrometer. The samples were prepared for analysis as glass discs to reduce the “matrix effect”.
The mineralogical composition of the untreated and treated samples was determined by X-ray diffraction (XRD), using a Bruker D8 Advance diffractometer (Bruker, Germany) with standard monochromatic Cu Kα radiation and operating at 40kV and 30mA. Scanning was performed with a 0.015° 2θ step size, and at 0.1s per step from 3° to 70°. Semi-quantifications were performing using the software Profex (version 3.14.0) [23].
The carbonate content of treated samples was determined by two analytical methods: differential thermal and thermogravimetric analysis (DTA-TG) and an elemental analyzer. DTA-TG were performed on a TG Netzsch STA 409 PC (Netzsch, Germany). Samples (around 150mg) were heated in an aluminium oxide crucible under a nitrogen atmosphere at 10°Cmin−1 from room temperature to 1200°C. Weight loss was measured by thermogravimetric analysis in the temperature range of 600–1000°C relative to the total carbonated decomposition. Elemental carbon content was measured using an elemental analyzer, Leco Truspec CHNS Micro (Leco, Michigan, USA), which calculated the carbonated ratio assuming that the whole carbon content was calcite.
The specific surface area (SSA-BET), micro- and nano-porosity were measured with an ASAP 2420 (Micromeritics, Georgia, USA) instrument using the adsorption of N2 at liquid nitrogen temperature (−196°C) and CO2 at room temperature. Before measurement, all samples were degassed at 150°C for 180min and finally outgassed to 10−3Torr. SSA was calculated using the classical Brunauer-Emmett-Teller theory (BET), and H2 and CO2 isotherms are analyzed using the Barrett–Joyner–Halenda (BJH) [24] method to yield micro- and nano- pore size distribution, respectively.
Macro- and meso-porosity were studied using a mercury porosimeter Pore Master 60-GT (Quantachrome Instruments Anton Paar, Austria). Low pressure mercury porosimetry applied to macropores while high pressure measured mesopores. The relationship between the pore diameter and Hg-intrusion pressure was calculated by the Washburn equation [25]. The pressures used in mercury porosimetry were from 1bar to 4000bar.
Soluble Ca, K, Na, Si and S ions were measured by a simultaneous inductively coupled plasma - optical emission spectrometry (ICP-OES) analysis using an ULTIMA 2 (Horiba Jobin Yvon, New York, USA) instrument. The specimens were prepared by mixing the solid powder samples with water and stirring for 24h, then concentrating the liquid phase by centrifugation followed by filtering through a Nylon 0.22μm syringe filter.
Micro-observations of morphological changes were obtained by scanning electronic microscopy (SEM) using a FEI Teneo (Thermo Fisher Scientific, Waltham, MA USA) microscope equipped with energy dispersive spectrometers (EDAX).
Due to the limits of detection and quantification of the different techniques used, the combination of analytical techniques allowed the qualitative analysis of the capture mechanisms and the indirect quantification of the amount of carbon captured [8].
Results and discussionSample characterization and selectionThe analysis of the majority elements of the samples analyzed (Table 2) shows an inverse relationship between the silicon and calcium content related to the compressive strength, being higher to greater SiO2.
Major oxide composition (wt%) by XRF of selected concretes.
| Sample | SiO2 | Al2O3 | Fe2O3 | MnO | MgO | CaO | Na2O | K2O | TiO2 | P2O5 | SO3 | LOI* |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hc1 | 34.49 | 3.74 | 1.77 | – | 5.46 | 27.34 | 0.37 | 0.74 | 0.16 | 0.07 | 0.96 | 24.45 |
| Hc2 | 23.74 | 3.80 | 2.24 | 0.04 | 2.24 | 35.51 | 0.74 | 0.60 | 0.27 | 0.05 | 0.53 | 29.25 |
| Hc3 | 6.62 | 0.52 | 0.31 | – | 0.28 | 50.13 | – | 0.14 | – | 0.03 | 0.47 | 40.09 |
| Hs1 | 69.35 | 6.64 | 2.87 | 0.05 | 0.57 | 12.29 | 1.24 | 1.14 | 0.27 | 0.14 | 0.77 | 5.14 |
| Hs2 | 70.61 | 5.17 | 2.18 | – | 0.50 | 9.88 | 0.97 | 0.91 | 0.20 | 0.07 | 0.48 | 8.26 |
LOI: Loss on ignition performed at 1025°C of furnace temperature.
The calcium oxide content in the concretes ranges from 9–50wt% and the magnesium oxide content ranges from 0.5–5wt%. Such high Ca and/or Mg materials seem to be appropriate for mineral carbonation.
The concretes, as it was expected, are mainly composed of quartz, carbonates and feldspars (alkali- and plagioclase). Minor phyllosilicates, gypsum and portlandite (calcium hydroxide) were also detected in some concretes (Table 3).
Mineralogical semi-quantification composition (wt%) of selected concretes. Abbreviations: Q – quartz; Ca – calcite; Do – dolomite; Alb – albite; Anor – Ca-felspar; Po – portlandite; Gyp – gypsum; Alu – alunite; Bi – biotite; Co – cordierite; I – illite; Ank – ankerite; Ett – ettringite; Cha – chamosite; Tr-trace (<5wt%).
| Sample | Q | Ca | Do | Alb | Anor | Po | Gyp | Alu | Bi | Co | I | Ank | Ett | Cha |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hc1 | 30 | 25 | 28 | 7 | 5 | Tr | Tr | Tr | Tr | – | – | – | – | – |
| Hc2 | 18 | 60 | – | 7 | Tr | – | – | Tr | – | Tr | – | 6 | – | – |
| Hc3 | 6 | 89 | Tr | – | – | – | – | Tr | – | – | – | – | – | – |
| Hs1 | 65 | Tr | – | 10 | 5 | 5 | – | – | – | – | 5 | – | 5 | Tr |
| Hs2 | 68 | 10 | – | 11 | Tr | – | – | – | – | – | Tr | – | – | Tr |
In limestone concretes the presence of carbonates (calcite, dolomite and ankerite) was predominant, varying from 53–89wt%, being these mainly the minerals rich in Ca (and/or Mg). Hc1 showed a percentage not much higher than 5wt% of other Ca-minerals that could be considered for carbonation (plagioclase and portlandite).
Siliceous concretes were mainly composed of quartz (65–68wt%). Hs2 presented 10wt% of calcite as main Ca-rich mineral, against Hs1 with a higher calcium content assigned to portlandite, plagioclase and ettringite (hydrated calcium aluminium sulphate) as possible minerals for carbonation.
For this reason, the siliceous concrete sample Hs1 was selected for carbonation tests in different particle size fractions, reaction times and different pressures.
Carbonation testsThe selected samples were tested for carbonation at room temperature, 20% humidity and 20bar CO2 pressure for different fractions and reaction times.
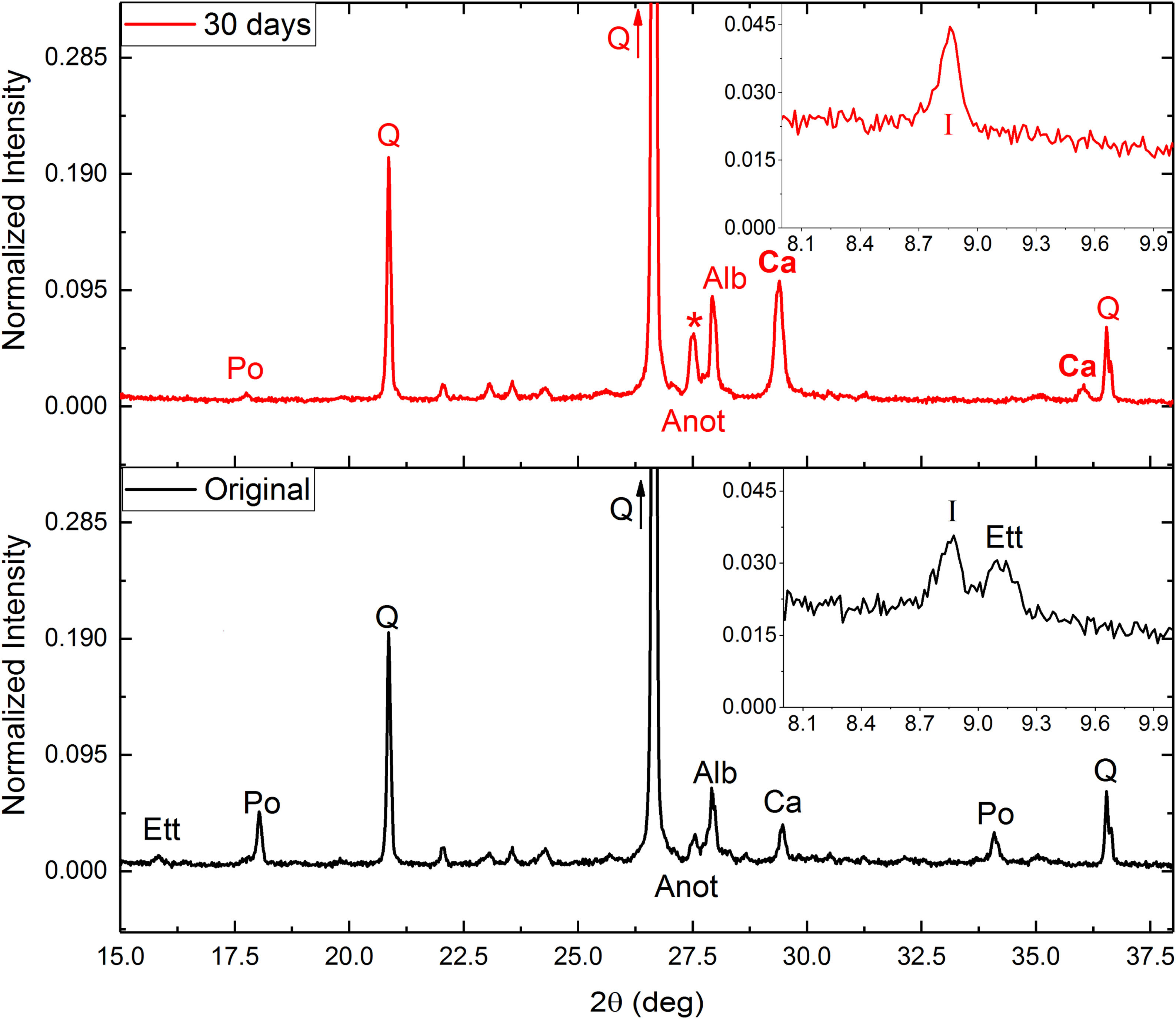
After these tests (in general) an increase in calcite intensity was observed by XRD (Fig. 2), as well as almost complete disappearance of the intensities corresponding to portlandite (Eq. (1)) and ettringite. The destruction of both minerals would provide calcium ions for the subsequent precipitation of the calcite, fixing CO2.
 and carbonated concrete (top) after 30 days and fraction 0.5–1mm. Po: portlandite, Q: quartz; Ett: ettringite: Anot: Anortite; Alb: Albite; I: Illite; Ca Calcite.")
Around 27.5° (asterisk in Fig. 2) a significant increase in intensity was observed. This increase could be due to a combination of two effects: (i) an increase in the calcium plagioclase content; and (ii) the appearance of a dehydrated phase of the ettringite due to drying of the samples after carbonation tests. This phase would have a structure equivalent to β-CaSO3[26]. Nonetheless, some ettringite should be destroyed due to an increase of silicon and sulfur ions compared with the original sample (Table 4). Also, an increase in free Ca ions were observed. Ca ions available for calcite precipitation due to the mineral destruction of portlandite and ettringite. Decrease in soluble K and Na may be related to increase in illite and albite respectively.
Soluble Si, S, Ca, K, and Na ions measured in untreated and treated after 30 days of reaction for Hs1 concrete.
| Sample | Pressure and temperature | Particle size fraction (mm) | Reaction time (days) | Si (mg/l) | S (mg/l) | Ca (mg/l) | K (mg/l) | Na (mg/l) |
|---|---|---|---|---|---|---|---|---|
| Hs1 | Original | 0 | 1.0 | 34.6 | 64.6 | 21.1 | 18.8 | |
| 10 bars25°C (room temperature) | 1–2 | 30 | 26.4 | 150.2 | 146.7 | 5.6 | 9.6 | |
| 1 | 26.8 | 159.3 | 164.5 | 5.5 | 11.2 | |||
| <0.5 | 28.1 | 171.6 | 178.1 | 3.7 | 5.1 |
While Eq. (1) showed the carbonation reaction of portlandite, the following equation corresponds to ettringite [27]:
According to TG results (Table 5), as quantification of CO2 fixed, were proportional to the reaction time and inversely proportional to fraction size. In other words, 30 days and <0.5mm fraction was the higher CO2 capture amount to Hs1, around 6.5wt%. However, this value was higher calculated from C-elemental as CO2, around 7.8wt% (an increase of 2.1wt% of C respect to the original value). The difference was due to some proportion of CO2 was physically retained (physical adsorption).
Loss mass (at 600–1000°C) by DTA-TG and C-elemental data of concrete Hs1 tested.
| Sample | Pressure and temperature | Particle size fraction (mm) | Reaction time (days) | DTA-TG Δm (wt%) | C-elemental (wt%) |
|---|---|---|---|---|---|
| Hs1 | Original | Total | 0 | 2.03 | 0.816 |
| 10 bars25°C (room temperature) | 1–2 | 1 | 2.13 | – | |
| 5 | 2.84 | – | |||
| 10 | 3.09 | – | |||
| 30 | 5.39 | – | |||
| 0.5–1 | 1 | 5.84 | – | ||
| 5 | 6.32 | – | |||
| 10 | 6.85 | – | |||
| 30 | 7.03 | – | |||
| <0.5 | 1 | 7.00 | 2.363 | ||
| 5 | 7.10 | 2.570 | |||
| 10 | 8.01 | 2.630 | |||
| 30 | 8.53 | 2.933 |
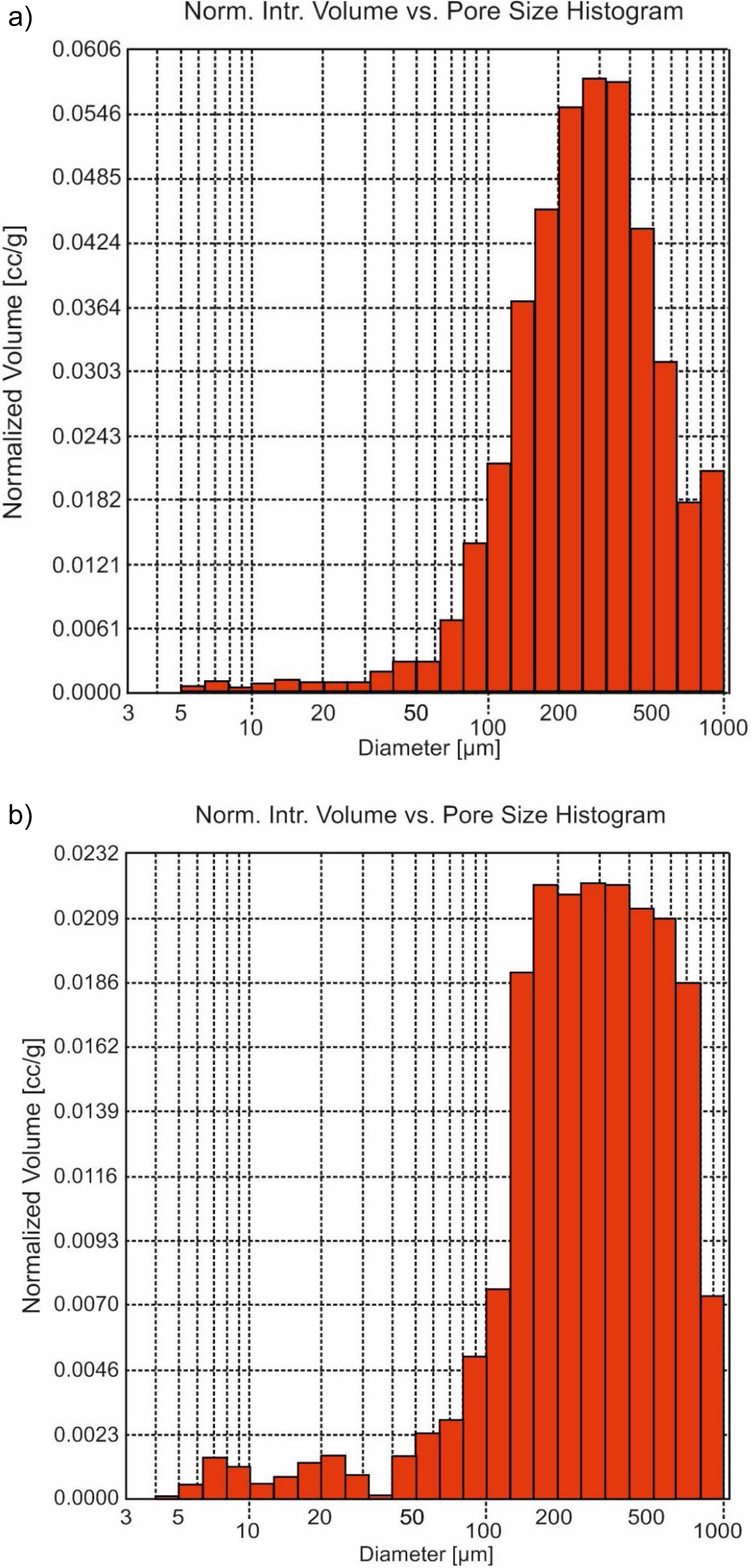
The morphological time evolution was analyzed using the minor fraction size (<0.5mm). Macro- and meso-porous were determined by Hg-porosimeter, which affects the proportion and size of the pores (Fig. 3). The maximum volume of this porous diameter was in the range around 150 to 650μm (Fig. 3b) in front of the untreated sample, which maximum was around 200–400μm (Fig. 3a). So, an increase of porous diameter was observed due to the acidic attack on the surface in the carbonation process [20].
 original and (b) treated Hs1 concrete at 30 days and <0.5mm fraction size.")
Whereas the smaller pore, micro- and nano-pore, determined by N2 and CO2 physisorption (Table 6) shown a decrease of the pore diameter to 5nm from around 15nm. This reduction was reached after 5 days of reaction, remaining constant at longer times. Combined with the decrease in the N2 and CO2 BET specific surface due to the smoothing of the irregular surface were related to the growth of calcite crystals that complete the pore by decreasing to smaller pore size as chemical CO2 retention.
N2 and CO2 BET specific surface area, and average (nano-/micro)-pore diameter for different reaction time Hs1, fraction <0.5mm.
| Sample | Pressure and temperature | Particle size fraction (mm) | Reaction time (days) | N2-BET SSA (m2/g) | CO2-BET SSA (m2/g) | Pore diameter (nm) |
|---|---|---|---|---|---|---|
| Hs1 | Original | Total | 0 | 59.27 | 72.77 | 14.72 |
| 10 bars25°C (room temperature) | <0.5 | 1 | 22.41 | 14.43 | 7.65 | |
| 5 | 29.79 | 15.30 | 5.27 | |||
| 10 | 28.48 | 15.93 | 5.41 | |||
| 30 | 33.38 | 16.87 | 5.22 |
Therefore, the mechanism would be as follows: (i) the mixture of CO2 and water, or CO2 in solution, begins to penetrate the empty pores; (ii) within the pore by the action of the acidity of the carbonic generated by the combination of gas and moisture, it dissolves calcium minerals (i.e. portlandite, ettringite); (iii) the released calcium ions combined with carbon ions precipitate as calcium carbonate filling the pores.
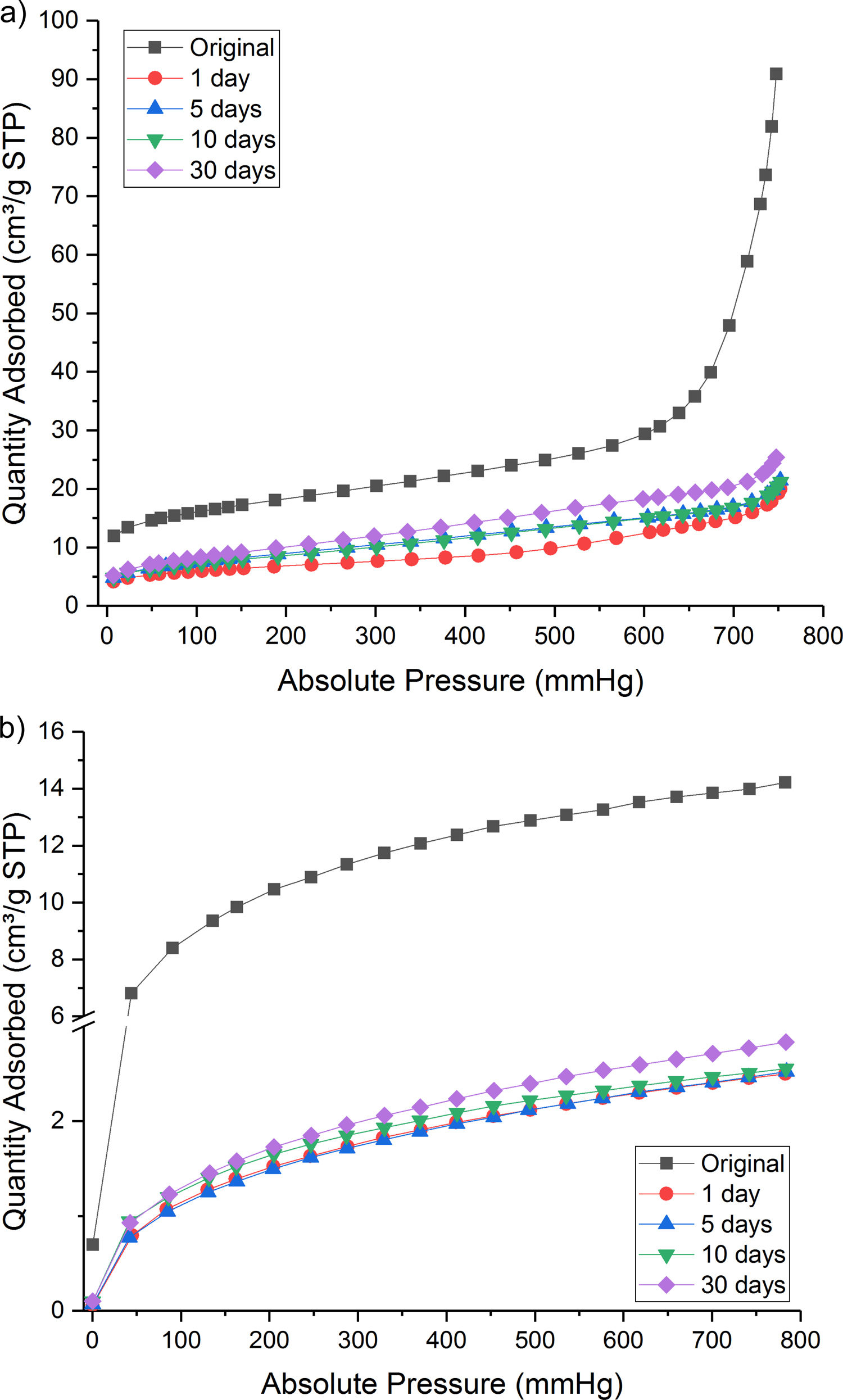
Nevertheless, some proportion of CO2 gas was physically adsorbed as shown in isothermal N2 and CO2 absorption curve (Fig. 4a and b). Both the treated samples presented a lower quantity of adsorbed gas than the original Hs1 concrete.
 N2 adsorption and (b) CO2 adsorption isotherms for original and treated Hs1 concrete at different reaction time and <0.5mm fraction size.")
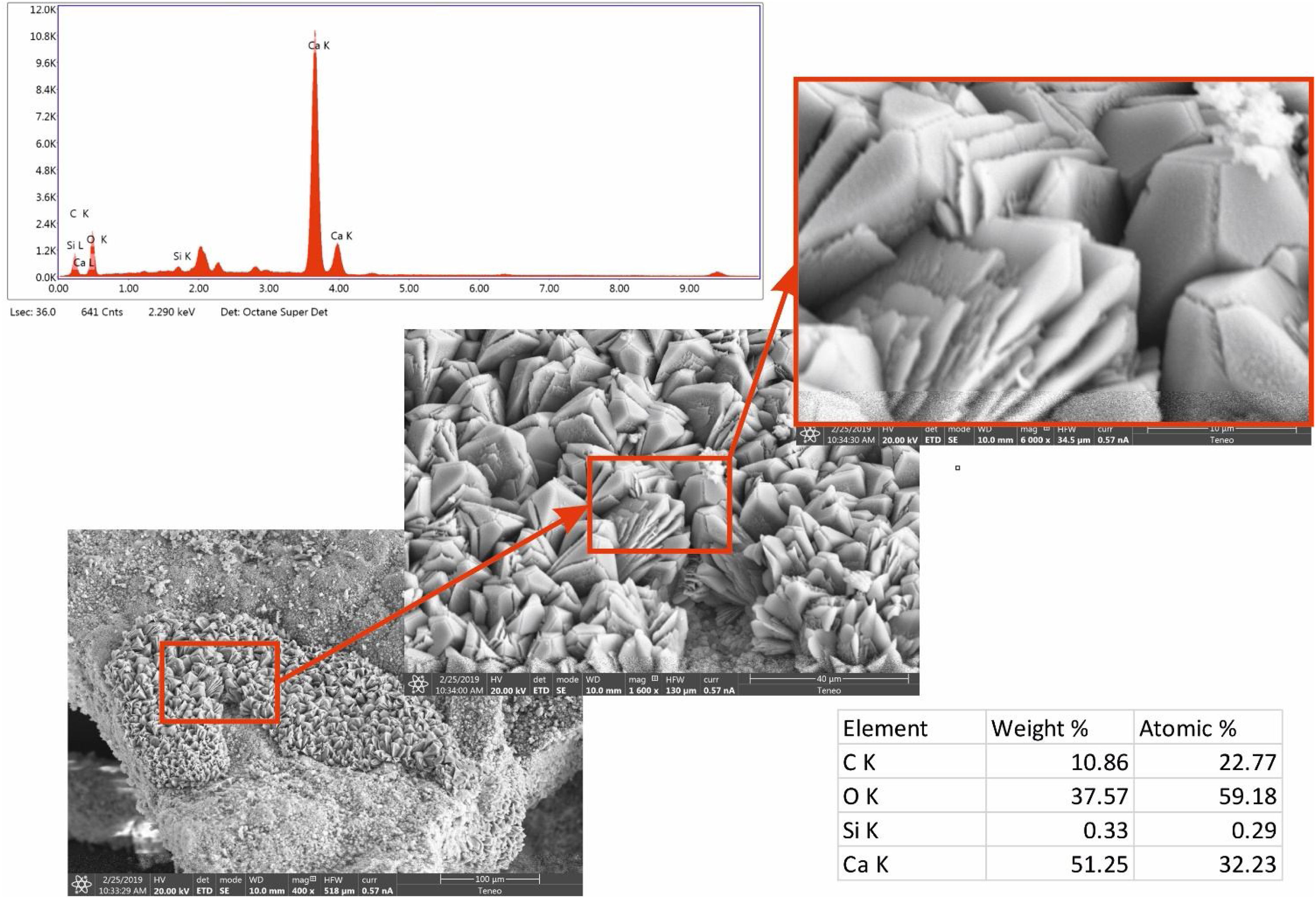
Micro observations confirmed the presence of precipitated calcium carbonate (Fig. 5). That neoformed calcite exhibits different habits. Mainly, geometric aggregates or with lamellar habit and dendritic growth were observed. Chemical analysis by EDS shows Ca, O and C (which was in the limit of proper quantification by the instrument) as principal elements. Also, a peak in the spectrum (not quantified) correspond to Pt, the metal used to the spattering.
 at 30 days reaction time.")
Some of these crystals were observed damaged, with cracks or fractures on the edges (Figs. 5 and 6). These alterations in the geometry of regular crystals may be due to the growth of new crystals and the destructive action of the acidic environment.
Conclusions
This work has studied the possibility of using concrete waste for mineral carbonation in a short time under viable conditions of low-medium pressure (10 bars), low relative humidity (20%) and ambient temperature.
Ca-rich minerals, as portlandite and ettringite, were partially dissolved and free Ca ions may be removed for subsequent carbonate precipitation, fixing the CO2 chemically. Some physical adsorption was also observed.
Compared to the bricks previously studied [4,8,20], in the concretes the main mechanism of CO2 capture is the physical absorption versus the mineral carbonation. Although in both types of components of C&D waste, both types of mechanisms were present.
The carbonation performance was higher at smaller grain size and higher reaction time. Around 6.5wt% retention of CO2 is achieved under favourable conditions of low pressure and temperature with low energy costs and therefore, environmental protection.
These results open the opportunity to use construction and demolition waste containing concrete for CO2 capture, in a full-scale recovery that would guarantee the profitability of the process.
FundingThis work was supported by the Junta de Andalucía [grant number P12-RNM-568].
This research was funded by the Andalucía Government (RDCCO2 project, P12-RNM-568), and the contract of Domingo Martín granted by the V Plan Propio de Investigación from the University of Seville (Spain). XRD, XRF, BET, C-elemental analysis, Hg porosimetry, SEM were performed using the facilities of the General Research Center at the University of Seville (CITIUS).

