










The current approach in bone tissue engineering requires resorbable biomaterials that enhance bone formation while maintaining sufficient mechanical stability. In this work, the influence of three levels of B-type carbonate substitution in hydroxyapatite lattice on mechanical strength and degradation rate is analyzed. The inverse aqueous route has been selected as a synthesis method of four powders with carbonate substitution between 4 and 6wt.%. X-ray fluorescence (XRF), (C-S)-Analysis, FT-Infrared, X-ray diffraction, DTA-TG and TEM were used to investigate chemical composition, type of substitution, thermal behaviour, and morphology of the powders. Disc shaped specimens were processed by uniaxial pressing and sintering in argon/CO2 flow. Maximum temperatures of thermal treatment between 750 and 850°C were selected to obtain similar porosity levels for the different compositions. The highest carbonate substituted material (5.3wt.%) presented higher compressive strength and dissolution rate than the other materials showing the beneficial effect of B-type substitution in hydroxyapatite materials for bone repair.
El enfoque actual en la ingeniería de tejido óseo requiere biomateriales reabsorbibles que promuevan la formación de hueso y conserven al mismo tiempo estabilidad mecánica. En este trabajo, se analiza la influencia de tres niveles de sustitución tipo B de carbonato en la red de cristalina de la hidroxiapatita, evaluando la resistencia mecánica y la tasa de degradación. Se ha escogido el método de síntesis por vía acuosa inversa para la síntesis de polvos con contenidos de carbonato entre 4 y 6% en peso. Se empleó fluorescencia de rayos X, análisis (C-S), infrarrojo TF, difracción de rayos X, TGA_DTA y MET para investigar la composición química, el tipo de sustitución, el comportamiento térmico y la morfología de los polvos. Se conformaron piezas en forma de disco mediante prensado uniaxial y fueron sinterizadas en flujo de argón/CO2. Se seleccionaron temperaturas de tratamiento térmico entre 750 y 850°C para obtener niveles de porosidad similares en todos los materiales. El material con mayor sustitución de carbonato (5,3% en peso) presentó mayores valores de resistencia a la compresión y velocidad de disolución que los otros materiales, lo que demuestra el efecto beneficioso de la sustitución de tipo B en materiales para la reparación ósea.
It is well known that bone-associated diseases due to ageing or traumas are one of the main problems of public health. In many cases, medical treatments consist on the implantation of temporary or permanent prosthesis, for which the used material is frequently an issue [1]. Biological materials – xenografts, human allografts and, above all, autografts – have been successfully used for a long time [2]. However, there are some problems associated such as limited amount of donor tissues, donor site morbidity, and/or potential risks of immunological incompatibility and disease transfer [3].
The alternative to biological materials is synthetic bone grafts which are made of different biocompatible materials depending on the required function. In addition to being biocompatible, materials for bone regeneration must be bioactive, promoting the formation of new bone tissue, and resorbable. The ideal material for bone regeneration would completely disappear once the new bone is grown [4,5].
In clinical practice, the most common materials used to fill bone defects are single phase or composite ceramics containing the two main inorganic constituents of bone, phosphorus and calcium together with other ions such as Mg or Si [6]. Main constituents of these ceramics are hydroxyapatite (HA, Ca10(PO4)6(OH)2) and beta-tricalcium phosphate (β-TCP, Ca3(PO4)2). Updated comprehensive reviews dealing with ceramics for bone regeneration are available [7–9].
The chemical composition of stoichiometric hydroxyapatite is similar to that of the mineral phase of bone and it is osteoconductive thus, it is widely used as bone regeneration material. However, it presents very limited resorbability which leads to the presence of residual synthetic HA in the healed bone defect after long times (several years) [9].
There are two basic approaches to tune the resorbability of hydroxyapatite for the fabrication of bone implants. One of them takes advantage of composite materials consisting of phases capable of dissolving at different rates, as in the work by S. Vanhatupa et al. [10]; the other one is based on ion substitutions in the crystal lattice of the ionic compounds. This second approach is widely used to improve the bioactivity of synthetic hydroxyapatite. Substitutions of (Ca2+), (PO43−) or (OH−) groups in the apatite structure by different ions generating distortions in the crystal lattice to improve its resorption rate have been investigated by different groups. Published results have been summarized in updated critical reviews [11–15].
The capability for the continuous bone regeneration in vivo partially relies on the non-stoichiometry of natural hydroxyapatites, in which relatively high amounts of CO32−≈3.5, 5.6 and 7.4wt.% for enamel, dentine and bone, respectively) and other cation and anion substitutions are found [16,17]. Thus, one of the most promising approaches for improving the bioactivity resorbability and osteoconductivity of hydroxyapatite is that of changing its composition towards that of bone hydroxyapatite by the substitution of hydroxyl (OH−) and/or phosphate (PO43−) ions by carbonate (CO32−) ions. Such materials are called carbonated hydroxyapatites (CHA) and are classified according to carbonate position in the crystal lattice of HA (Ca10(PO4)6(OH)2). In A-Type CHA, hydroxyl ions are replaced by carbonate ions while in B-type, carbonate ions are in phosphate sites; both types of substitution are also possible, and this apatite is known as AB-type [18,19].
B-type is the preferential carbonate substitution found in bones of most species, including the human one; the chemical compositions of biological apatite vary widely among the different bones and individuals [20]. The replacement of the large tetrahedral phosphate ions by the small planar carbonate ions leads to changes in lattice parameters, namely, a decrease a-axis and an increase c-axis. Such distortion of the crystal structure results in a decrease in the stability of the apatite structure, resulting in increased reactivity as discussed below [12].
CHA presents a higher dissolution rate in weak acidic conditions (pH=3.5) than HA [13,21], which explains the higher osteoclastic osteoconductive and resorption capabilities t of CHA as compared to HA [22,23]. At the physiological pH (∼7.4), both hydroxyapatites present similar dissolution rate [24]. Accordingly, several authors have demonstrated the bioactivity of CHA to be higher than that of HA in vitro [25,26] and in vivo [27–30]. Moreover, it has been suggested that the amount of carbonate might modulate the biological behaviour, i.e., osteoclast and osteoblast responses, as highlighted by Ortali et al. [31]. In this sense, Miyamoto et al. have demonstrated the effectiveness of carbonate apatite in the shape of granules [29,30,32] blocks [27,33] for bone regeneration. Preliminary results of the effectiveness as bone substitute of the high carbonate content (12.0wt.%) apatite developed by these authors as compared to those of other two commercially available materials (with 0.1 and 5.5wt.% of carbonate) suggest that CO32− content may be one of the factors governing bone formation and resorption [27]. Nevertheless, although the histological comparison reported by these authors is important, there were also dissimilarities between the three analyzed commercial products other than CO32− contents, such as crystallite size, surface morphology, and method of fabrication that might also influence their bioactivity.
Spence et al. [26] by comparing different specimens with carbonate contents from 0.1 to 2.35wt.% concluded that resorption rate of CHA in the presence of human osteoclasts increased with increased carbonate content.
Murugan et al. [33] studied resorption of CHA in simulated physiological conditions, they evaluated the weight loss of CHA pellets (2wt.%) in phosphate buffered saline solution (pH=7.2) during 800h, finding that, after this period, CHA presented a higher weight loss (95wt.%) than stoichiometric HA (90wt.%) indicating higher dissolution rate due to carbonate addition. It has been proposed that the decrease in the lattice crystallinity associated to the presence of B-type substitution is responsible for the increase in solubility of CHA in both in vitro and in vivo tests [34].
Mechanical performance is not a main issue in the case of materials for bone regeneration as it is in the case of permanent implants. However, bone regeneration scaffolds should exhibit sufficient initial mechanical strength and stiffness to substitute for the mechanical function of the damaged bone until it is regenerated and/or permit cell seeding of the scaffold in vitro without compromising scaffold architecture [35]. Therefore, the influence of carbonate substitution on mechanical behaviour of hydroxyapatite is one of the main aspects to be considered. Some authors have reported relationships carbonate-mechanical properties; however, it does not exist a systematic study in the literature on increasing levels of carbonate substitution on the mechanical behaviour of hydroxyapatite [36,37]. The available works report results of mechanical performance of CHA materials with specific carbonate contents. Bang et al. [38] reported tensile strength values determined by the diametral compression test of dense specimens. These authors found that the strength of the CHA material with 11wt.% CO32− substitution (≈11MPa) was about double of that of the stoichiometric HA one (≈6MPa). Regarding porous (45% porosity) scaffolds, Landi et al. [34] reported values of compressive strength for CHA with 8.8wt.% of carbonate content (6.0±0.5MPa) that were twice as much as those for HA (3.1±0.3MPa).
In the same way, Zhu et al. [39] found the carbonated hydroxyapatite to have higher flexure strength than unsubstituted HA due to the dense and uniform fracture surface without obvious cracks.
From the above discussion it can be concluded that the capacity of B-type carbonated hydroxyapatite to be replaced by bone is tightly linked to its resorption capability, thus, to the level of carbonate at the phosphate sites of the crystal lattice. Such a substitution also improves the mechanical strength of CHC. However, the relative importance of compositional parameters on the degradation and mechanical behaviours are still unclear. To develop CHA scaffolds with adequate properties, it would be necessary to establish the effect of systematically varying levels of CO32− substitution on the mechanical and physiological behaviours of hydroxyapatite materials with similar microstructural features.
The behaviour of materials once implanted in the body can be predicted by using in vitro accelerated and/or long-term tests. In the case of HCA, accelerated degradation studies can be done in acidic medium to mimic the osteoclastic environment (pH<3) required for resorption [40]. To study the long-term (few weeks) degradation of materials at physiological conditions simulating the body pH it would be necessary to use buffer solutions to avoid the formation of an apatite layer that would arrest the dissolution process [41].
There have been proposed different methods to synthesize CHA powders either at room or high temperature. Molar ratios Ca/P=1.67 are required to avoid the formation of secondary phases like tricalcium phosphate (TCP) and calcium oxide (CaO) [42]. One alternative for obtaining CHA is the extraction of calcium phosphate from natural sources such as mammalian bones, aquatic species, plants and minerals [17]. In general, long processing times are needed for such extraction, which makes it energy intensive, and the morphology of the particles cannot be controlled even at high temperature. Solid state reaction of CaCO3 and Ca3(PO4)2 at 900°C in dry CO2 atmosphere was used by Walley et al. [43] to obtain A-type CHA. In the hydrothermal method [44], [45], a precursor suspension is autoclaved at temperatures between 120 and 290°C. This process is often used to prepare nanocrystalline HA with controlled size and morphology; its main disadvantage is the difficult industrial scaling. The acid–base neutralization reaction, which uses calcium hydroxide (Ca(OH)2), phosphoric acid (H3PO4) and CO2 gas, does not permit the accurate control of substitution level. In addition, this process involves a high probability of CO32− ions remaining adsorbed on the surface of the crystals, thus, being easily lost during heat treatment [37]. Finally, the precipitation synthesis consists in mixing calcium, carbonate and phosphate suspensions prior to precipitating the homogeneous mixture. This method is classified according to the order in which the precursor suspensions are mixed. In the direct route [18,31] a precursor suspension of carbonate and phosphate is held in the reactor while a calcium precursor suspension is added dropwise. In the inverse route, a carbonate and phosphate precursor suspension with controlled CO32−/PO43− molar ratios is added dropwise to a calcium phosphate suspension held in the reactor. This later method is preferred for the synthesis of B-type CHA, due to its simplicity and its capability for controlling the main characteristics of the synthesized powder: carbon content, grain size, granulometric distribution, morphology and specific surface [18,44,46,47].
The aim of this study was to determine the effect of carbonate contents in the range of those of natural hydroxyapatite on compressive strength and in vitro degradation of B-type carbonated hydroxyapatite materials with similar microstructural parameters. The inverse aqueous route was selected as synthesis method due to its potential to control the main characteristics of the product by carefully monitoring the synthesis parameters. Adequate sintering schedules for the different carbonate contents have been established in order to reach materials with similar levels of porosity to allow the determination of the sole effect of the carbonate content on the material behaviour.
ExperimentalSynthesis and characterization of the powdersAqueous solutions of calcium nitrate tetrahydrate [(Ca(NO3)2)·4H2O, Merck®, Germany], ammonium hydrogen phosphate [(NH4)2HPO4, Merck®, Germany] and hydrogen ammonium carbonate [(NH4)HCO3, Alfa Aesar®, USA], were used as reactants. In order to reach the envisaged carbonate contents in the final materials (4–6wt.%), to prevent the formation of calcite (CaCO3) and to obtain powders with reproducible characteristics, the adequate amounts of solutions to hold a molar ratio Ca/P=1.67 for all syntheses and were used. The substitution level was designed according to molar ratios CO32−/PO43−=0, 0.25, 0.375, 0.5; the associated number of moles of carbonate (ncarbonate) used for the synthesis of powders HA, C2, C3 and C4, respectively are listed in Table 1. First, the solution of calcium nitrate tetrahydrate, with pH=9 controlled by the addition of NH4OH (Merck®, Germany), was stirred in a reactor at 90°C in argon atmosphere. Then, the adequate amounts of solutions of ammonium hydrogen phosphate and hydrogen ammonium carbonate were added dropwise, and the mixed solution was ripened during 30min, filtered, and washed with deionized water following the method described by Lafon et al. [48]. The precipitated material was dried at 80°C for 24h and heat treated for 2h at 400°C in air to remove synthesis residues as nitrous species, finally powders were passed through a 45μm sieve.
The chemical composition (Ca/P molar ratio) of the powders was determined by wavelength dispersive X-ray fluorescence (WD-XRF ARL Optim X ThermoFisher, USA). The carbonate content (wt.% CO32−) was estimated from the carbon content determined by elemental analysis (C–S test analyzer; Leco, USA), given values are the average of 5 measurements and errors are the standard deviation. The type of carbonate substitution in apatite was determined by Fourier transform infrared spectroscopy with (FTIR, IR Tracer, Shimadzu, Japan). The crystalline phases were determined by X-ray diffraction (Empyrean, PANanalytical, B.V., The Netherlands), using CuKa (λ=1.540598Å) radiation, in the range of 10–60° with 50s and 0.02° step and Standard International Centre for Diffraction Data (ICDD) card no. 9-0432 for HA. The Rietveld refinement was done with software Xpert Highscore 2.0, (PANanalytical, B.V., The Netherlands). The refinements were based on the literature data using the hexagonal crystal (space group P63/m) for HA with lattice parameters a=b=9.4219Å and c=6.8822Å, according procedure of Ezekiel [34,49].
The morphology of the HA particles and those of the powder with the maximum carbonate content was observed by transmission electron microscopy (TEM, FEI Tecnai 20 G2 S-Twin 200kV, FEI, USA).
Thermogravimetric and differential thermal analysis, TGA/DTA (TGA & DTA/DSC thermal analyzer, Setsys Evolution, Setaram, France) with a heating rate of 10°C/min up to 1300°C were performed to analyze the thermal stability in air of the materials.
Preparation and characterization of the sintered materialsCylindrical green compacts were fabricated by uniaxially pressing (40MPa) using a steel die of 6.3mm internal diameter in a manual press (Lordeco, Colombia). The heights of the cylinders were ≈4mm for chemical characterization and degradation studies and ≈13mm for mechanical characterization.
Sintering was performed in an electrical tube furnace (Carbolite 3216, UK), in Argon/CO2 (50/50vol%) flow (5mL/min) and using 5°C/min as heating and cooling rates and 2h holding time at maximum temperature. The specific maximum sintering temperature for each composition was selected to obtain similar porosity levels for all materials.
Zyman and Tkachenko [50] have reported transition from B to A substitution during thermal treatments in CO2 atmosphere at temperatures above 800°C depending on the carbonate content. Lafon [51] established that an atmospheric flux of Ar/CO2 mixture and maximum temperature of 800°C avoided A substitution in the CHA powders. Therefore, maximum sintering temperatures lower than 800°C were selected for the three CHA powders First, the temperature was set as 750°C for composition C4, which is most sensitive to carbonate loss during heat treatment. This temperature was enough to reach sintered C4 compacts with sufficient structural integrity and high open porosity ≈50%. Then temperatures of 850, 780, 780 and 750°C for HA, C2, C3 compacts, respectively, were selected to reach similar levels of open porosity. Final dimensions of the sintered specimens were diameter=6.0±0.2mm and heights=3.5±0.1mm and 12.5±0.2mm for chemical and mechanical characterizations, respectively.
Open porosity of sintered specimens was determined following EN 1389:2003. Given values are the average of 5 measurements and errors are the standard deviations.
The crystalline phases and the type, and content of carbonate substitution in the sintered specimens were characterized following the same procedures as for the synthetized powders (Section 2.1), by X-ray diffraction, FTIR and (C-S) analyses respectively.
The structural integrity of the “as sintered” cylinders was characterized by uniaxial compression following the ASTM C1424 standard in a universal testing machine (Shimadzu AGS-X 50kN, Japan) with steel plates and a rate of displacement of the load frame of 0.5mm/min. Given values are the average of 5 determinations and errors are the standard deviations.
Degradation studiesTwo different methods were used to investigate the degradation behaviour of the sintered materials. In both cases the degradation level was characterized as the difference between the weights of the specimens before and after the degradation tests. Prior to testing, the specimens were dried at 120°C overnight until a constant weight was achieved. An analytical balance Sartorius CP324S with precision of 0.1mg was used for all determinations. For each composition and each test, three nominally identical specimens were tested in the same conditions. The weight losses of three specimens were determined; given values are the average of the three determinations and errors are the standard deviations.
Accelerated degradation was performed using the procedure proposed by Diez-Escudero et al. [40], by immersing the specimens in an acidic solution consisting of 0.01M hydrochloric acid (HCl, Merck®, Germany) and 0.14M sodium chloride (NaCl, Merck®, Germany) at 37°C. Since the solution was not buffered, the samples were immersed in a large volume of acidic medium (15mL) in sterile polypropylene tubes (50mL) and every hour they were transferred to new vials with fresh medium. In this way, changes in the solution pH that would alter the degradation behaviour were prevented. After 8h immersion, the specimens were rinsed twice with distilled water and dried overnight at 120°C until constant weight.
For degradation in a physiological environment up to 7 weeks, specimens were first weighed, and their dimensions were measured using a precision calliper, to estimate their external surface. For testing, the three specimens were immersed in the Tris–HCl solution (pH 7.4, 37°C) with a ratio (specimen external surface/solution volume)>0.1cm−1 to prevent precipitation over long periods of time. The solutions were renewed after 1, 3 and 7 days and then after each week until reaching 7 weeks, according to Juraksi et al. [52]. The specimens were taken out at the scheduled time points, rinsed with deionized water three times, dried, weighed and immersed again. Statistical significance with a level of p<0.05 was evaluated and One-way ANOVA was used, as it is recommended for experimental results that obey normal distributions.
Results and discussionSynthesis of the powdersThe Ca/P molar ratios and the final carbonate contents for the synthetized powders are collected in Table 2. They are in the range of the expected compositions and values found in biological bone for the three carbonate powders (Ca/P=1.4–2 and CO32−=4–8wt.%), the Ca/P ratio varies according to the inclusion of carbonate in phosphate positions within the HA lattice [9]. According with the proportions of reactants used for the synthesis, increasing carbonate contents are accompanied by increasing Ca/P molar ratios, which would reveal that carbonate ions substitute the phosphate (PO43−) ones. The C2 powder showed higher relative efficacy in incorporating carbonate relative to the powders with higher carbonate content, as evidenced by the correlation between the molar quantity of carbonate in the powders (Table 2) and reactants (Table 1). This could be attributed to the lower amount of carbonate which would enhance the inclusion of the ion within the hydroxyapatite lattice.
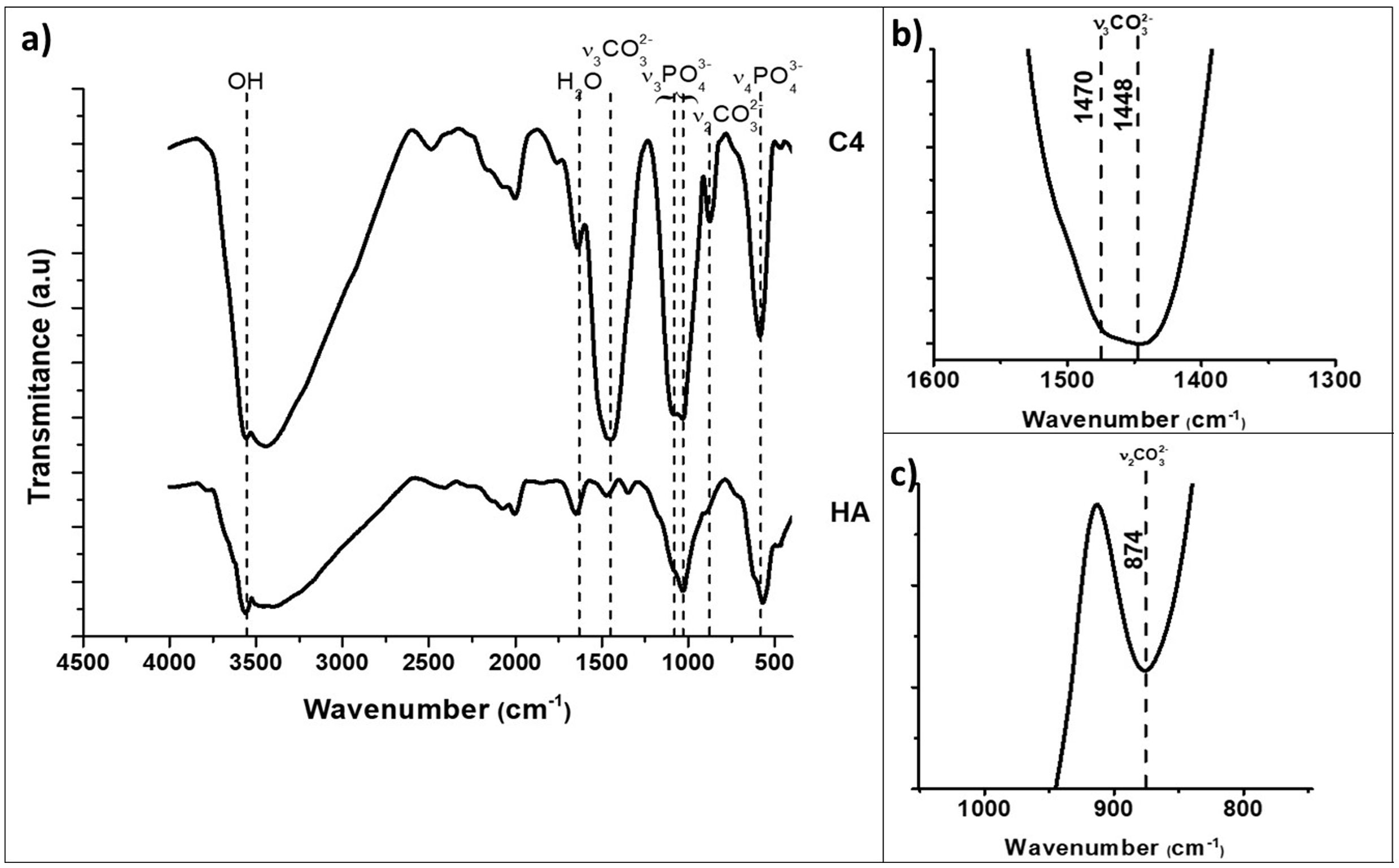
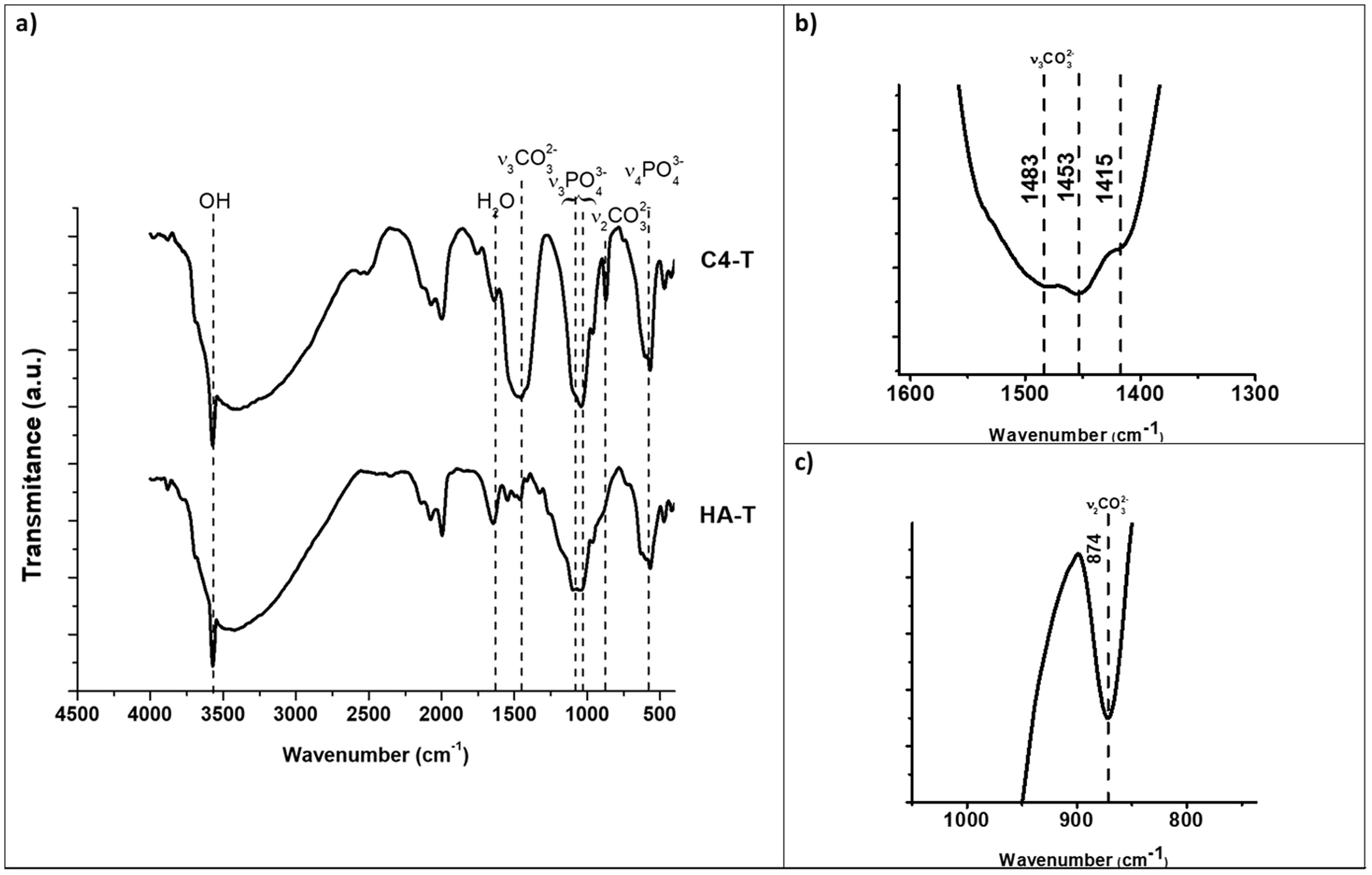
The kind of carbonate substitution in apatite is most readily detected by FTIR [1]. The FTIR spectra of the synthesized HA and C4 powders are shown in Fig. 1. The characteristic bands of the functional groups of hydroxyapatite are highlighted in Fig. 1, OH (3570cm−1), PO43− (550–570cm−1 for ν4 and 1020–1120cm−1 for ν3). The wavenumbers of vibrations modes ν2 CO3 and ν3 CO3 for composition C4 are summarized in Table 3 together with those corresponding to biological and synthetic substituted apatites [49]. The wavenumber values of the bands ν2 and ν3 associated to carbonate substitutions in the spectrum of C4 powder confirm the only B-type substitution in this carbonate hydroxyapatite, results for C2 and C3 powders are similar, due to this, only spectra of the powder with the highest carbonate content are presented.
 and highest substituted CHA (C4). The characteristic bands of the functional groups of hydroxyapatite are presented, OH (3570cm−1), PO43− (550–570cm−1 for ν4 and 1020–1120cm−1 for ν3). (a) Complete, (b) v3CO3 region (1600–1300cm−1), (c) v2CO3 region (900–800cm−1) zoom in C4 spectra.")
FTIR spectra of synthesized hydroxyapatite powders: unsubstituted (HA) and highest substituted CHA (C4). The characteristic bands of the functional groups of hydroxyapatite are presented, OH (3570cm−1), PO43− (550–570cm−1 for ν4 and 1020–1120cm−1 for ν3). (a) Complete, (b) v3CO3 region (1600–1300cm−1), (c) v2CO3 region (900–800cm−1) zoom in C4 spectra.
Wavenumbers (cm−1) of vibrations modes ν2CO3 and ν3CO3 characteristic ones for biological and synthetic substituted apatites [39].
| Experimental | Reference | ||
|---|---|---|---|
| C4 | CO3 in B-site | CO3 in A-site | |
| ν2 CO3 | 874 | 873 | 878 |
| ν3 CO3 | 1448 | 1445 | |
| 1470 | 1470 | 1542 | |
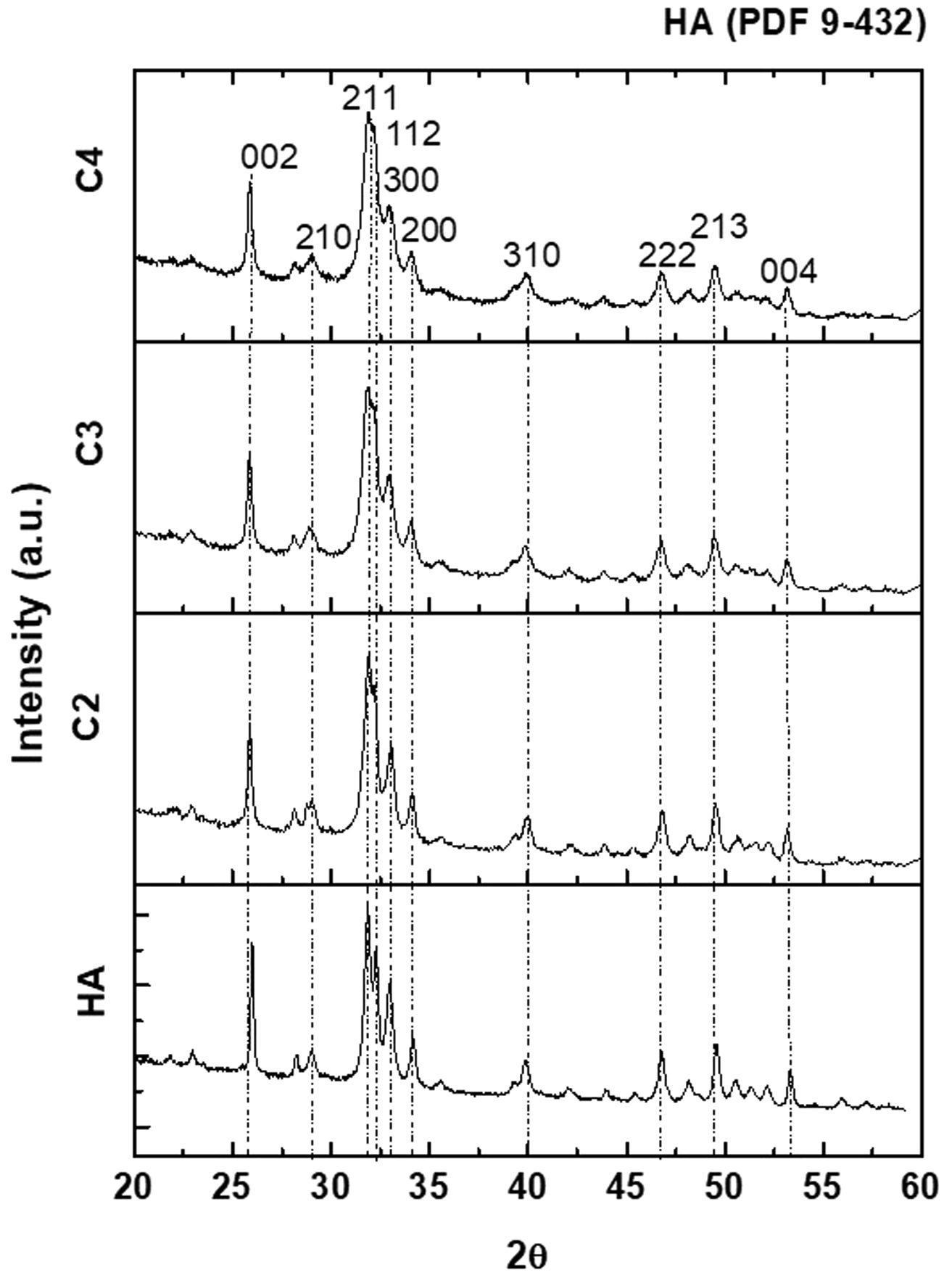
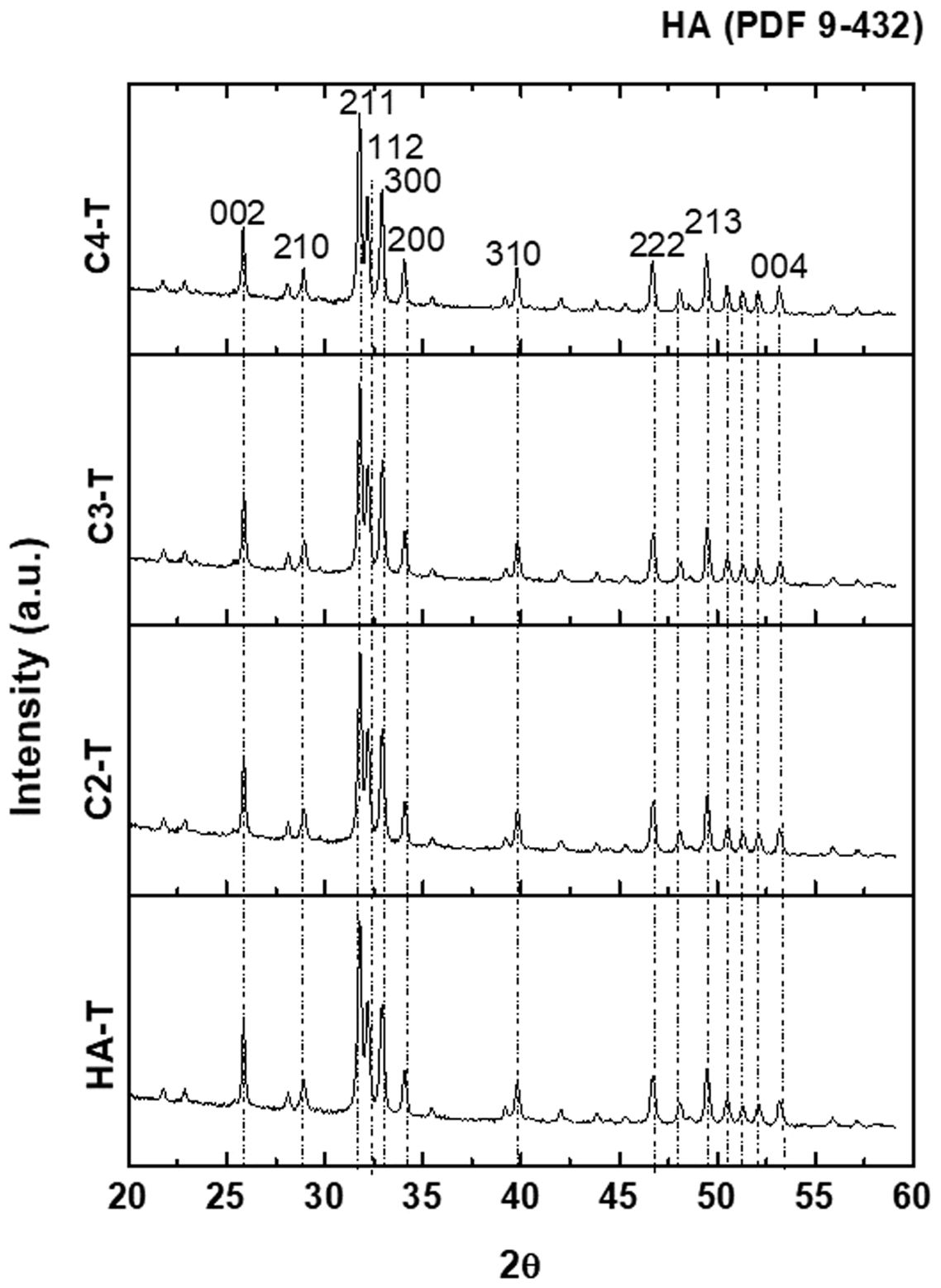
The XRD patterns of the four synthesized powders are plotted in Fig. 2; all identified peaks correspond to HA without secondary phases.

Lattice parameters from Rietveld refinement for the synthesized powders as a function of the carbonate content are shown in Table 4. For both parameters, a and c, differences between the values corresponding to the different compositions are statistically significant. Parameter a decreases as the carbonate content increases while parameter c follows the opposite trend, slightly increasing with carbonate content. These XRD results agree with the B-type substitution resulting in a progressive decrease of a parameter and a marginal increase of c due to the smaller size of the carbonate ion compared to that of the phosphate one, as reported by other authors [19,49,53]. Nevertheless, a straightforward relationship between the values of the a and c parameters and the B-type carbonate content does not exist because the deformation of the apatite lattice due to substitution by B-type carbonate varies markedly with the preparation method, the contents of A-type carbonate and other lattice ions, and minor amounts of cations replacing calcium [42,54].
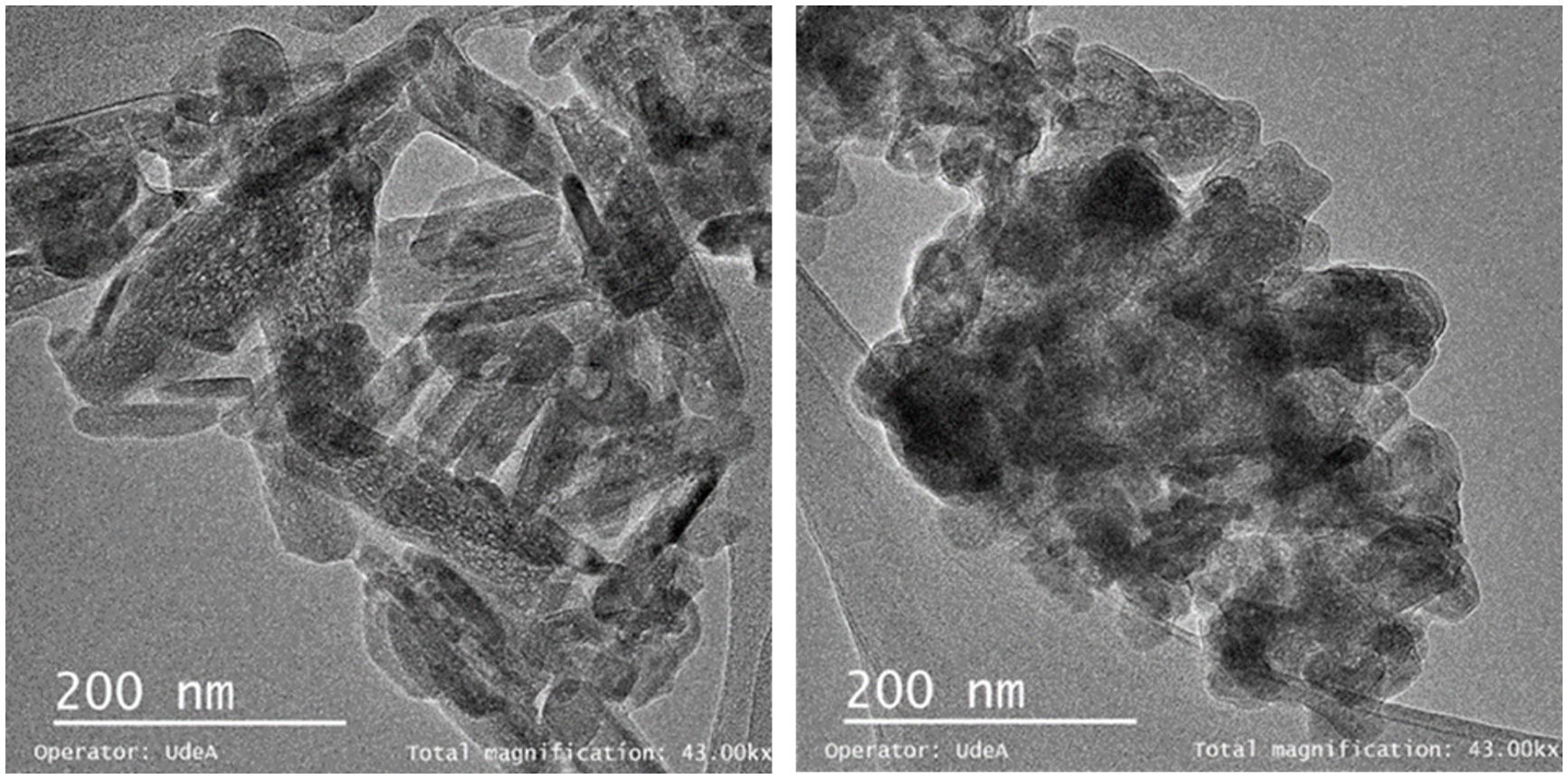
The TEM images of the “as synthesized” HA and C4 particles are shown in Fig. 3. The HA powder (Fig. 3a) consists of rod-like particles like those of the commercial powders [54]. The morphology of the C4 particles is equiaxial (Fig. 3b); such a change has been attributed to a change in crystal growth due to the alteration of the lattice parameters caused by phosphate replacement [53].
 HA, Rod-like particles, (b) C4, Equidimensional particles.")
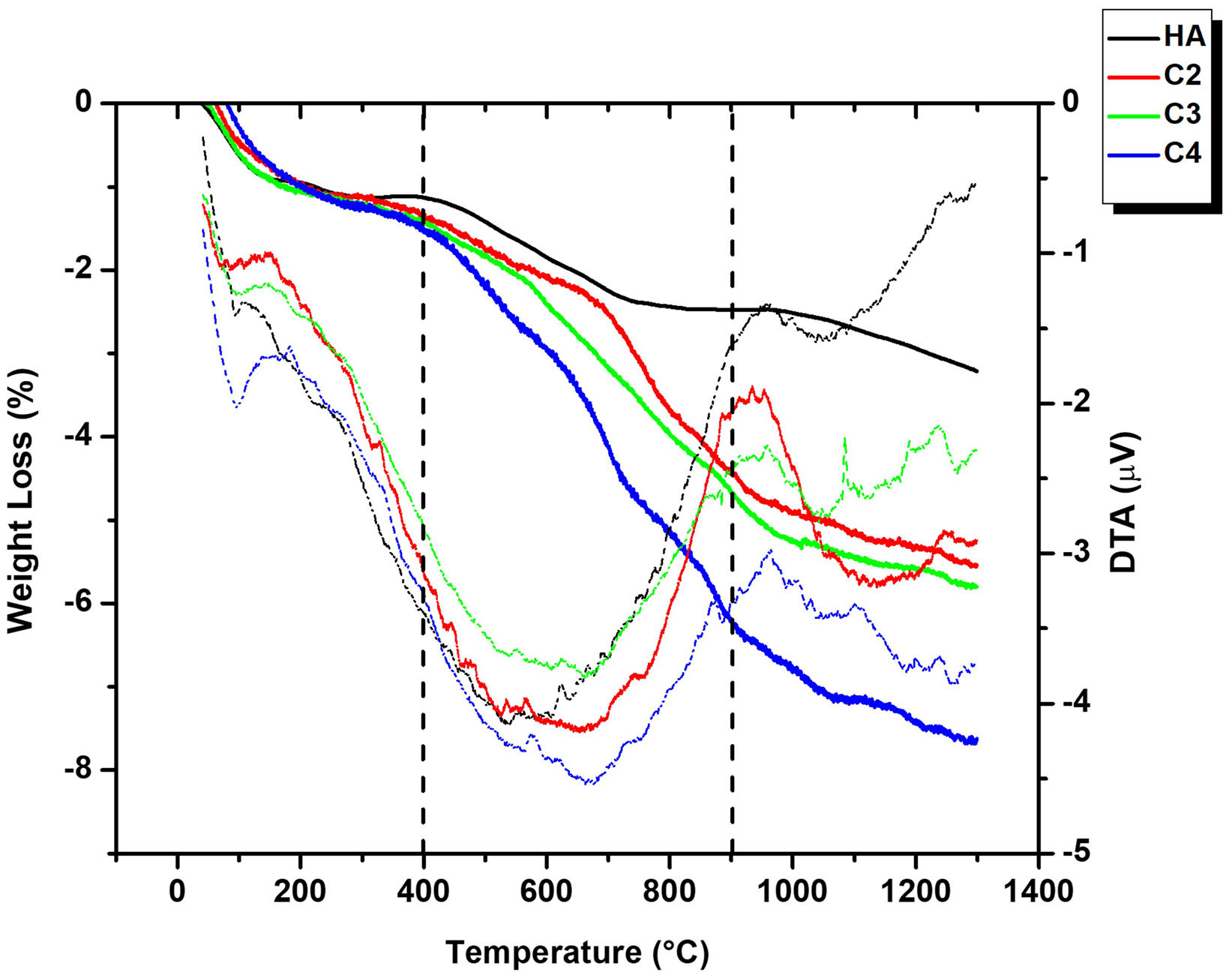
The results of the TGA/DTA in air for the four powders are presented in Fig. 4. Three stages can be identified as a function of the temperature interval. From room temperature to 400°C all powders showed a gradual mass loss attributed to the loss of adsorbed and lattice water [53]. In the second stage (400–900°C) a sharp weight loss, accompanied by a wide endothermic band observed in DTA, takes place due to carbonate decomposition [55]. This feature is the most extreme in the C4 curve due to its highest carbonate content (5.9wt.%). In fact, for all powders, values of the weight loss between 400 and 900°C, determined by TGA are like those of the carbonate contents inferred from the carbon determined by elemental analysis collected in (Table 2). In the final stage (900–1300°C), the weight loss is caused by dehydroxylation process [56]. The release of carbonate from temperatures as low as those of the second stage confirms the requirement of a controlled atmosphere for the sintering of CHA materials.
Characterization of the sintered materials, carbonate decomposition (400–900°C) and dehydroxylation process (>900°C).")
There is a slight decrease of the average values of the carbonate contents in the sintered materials (Table 5) as compared to those in the powders (Table 2), even though values are always inside the variability limits. The fact that carbonate is mostly kept in the materials after the sintering process demonstrates the adequacy of the sintering temperatures and atmosphere.
Porosity determines the strength values of materials with large pores. Therefore, materials with similar porosity levels are needed to determine the effect of carbonate content on strength. Previous systematic studies on the effect of sintering temperature on the open porosity, which was the major porosity-type in the sintered specimens, allowed the selection of sintering temperatures in the range 750–850°C to reach similar levels of open porosity (50–60%, Table 5) that would not mask the compositional effect on strength.
The XRD patterns of the sintered specimens are collected Fig. 5. For all materials, the identified peaks are the same as those of the synthesized powders (Fig. 2). For the sintered specimens, the peaks are sharper, which could be associated with a higher crystallinity due to the thermal treatment.
 while peaks are sharper.")
XRD diffraction patterns of sintered specimens. The identified peaks are the same as those of the synthesized powders (Fig. 2) while peaks are sharper.
Results of the Rietveld refinement of the lattice parameters for the sintered materials are summarized in Table 6.
For each composition, the value of the parameter a of the sintered material is significantly higher than that of the corresponding powder (Table 4) while c remains the same for low carbonate contents and decrease slightly for composition C4 after the sintering process. Other authors have reported that, as the treatment temperature increases, the parameter a exceeds the theoretical value of HA, due to the appearance of the substitution A-type [50,57]. However, such substitution was not evidenced in this work, thus, the changes in lattice parameters with the sintering treatment should be due to other processes such as increased crystallization, as revealed by the sharper XRD peaks (Fig. 5), and/or the small carbonate losses discussed above.
Fig. 6 shows the FTIR spectra of the sintered unsubstituted hydroxyapatite material (HA-T) and that with the highest carbonate substitution (C4-T). The observed bands are the same as those observed in the spectra of the corresponding powders (Fig. 1), which reveals that only B-type substitution is present in the sintered substituted material, C4-T, an additional slight band appeared at 1415cm−1, which has been reported by other authors in type B substitutions after thermal treatment [50]. The absence of A-type substitution has been assured by the selection of the sintering atmosphere and temperatures.
 and highest substituted HCA (C4). The characteristic bands of the functional groups of hydroxyapatite are presented, OH (3570cm−1), PO43− (550–570cm−1 for ν4 and 1020–1120cm−1 for ν3). (a) Complete, (b) v3CO3 region (1600–1300cm−1), (c) v2CO3 region (900–800cm−1) zoom in C4-T spectra.")
FTIR spectra of sintered hydroxyapatite specimens: unsubstituted (HA) and highest substituted HCA (C4). The characteristic bands of the functional groups of hydroxyapatite are presented, OH (3570cm−1), PO43− (550–570cm−1 for ν4 and 1020–1120cm−1 for ν3). (a) Complete, (b) v3CO3 region (1600–1300cm−1), (c) v2CO3 region (900–800cm−1) zoom in C4-T spectra.
The compressive strength values are shown in Table 5 together with the open porosity and carbonate content. The microstructures revealed by the fracture surfaces are collected in Fig. 7. All the microstructures, with high levels of open porosity, were characteristic of initial sintering stages [58,59]. In all cases, particles were submicronic and their size decreased from around 200nm in the unsubstituted material (HA, Fig. 7a) down to less than 100nm in both carbonated materials with the highest carbonate contents (C3-T and C4-T, Fig. 7c, d). No singular defects were identified as fracture origins in any fracture surface. Thus, the largest microstructural feature which is the open porosity, similar in the four materials, was responsible for fracture initiation, and it is possible to infer the sole effect of carbonate content on strength. Even though the average value of strength of C2-T was higher than that of HA-T (Table 5), however, differences are not statistically significant, which indicates that, the potential effect of the smallest carbonate content considered is masked by microstructural variability. Contrarily, strength values of C3-T and C4-T are significantly higher than those of HA-T, showing the beneficial effect of carbonate substitution on strength, increasing with carbonate content. The strength increase is due to the intrinsic effect of carbonate in the crystalline structure discussed above.
 tested in uniaxial compression. Similar high levels of open porosity are observed in all micrographs. (a) HA-T, (b) C2-T, (c) C3-T, (d) C4-T.")
FE-SEM micrographs of Au coated fracture surfaces of sintered specimens (Table 5) tested in uniaxial compression. Similar high levels of open porosity are observed in all micrographs. (a) HA-T, (b) C2-T, (c) C3-T, (d) C4-T.
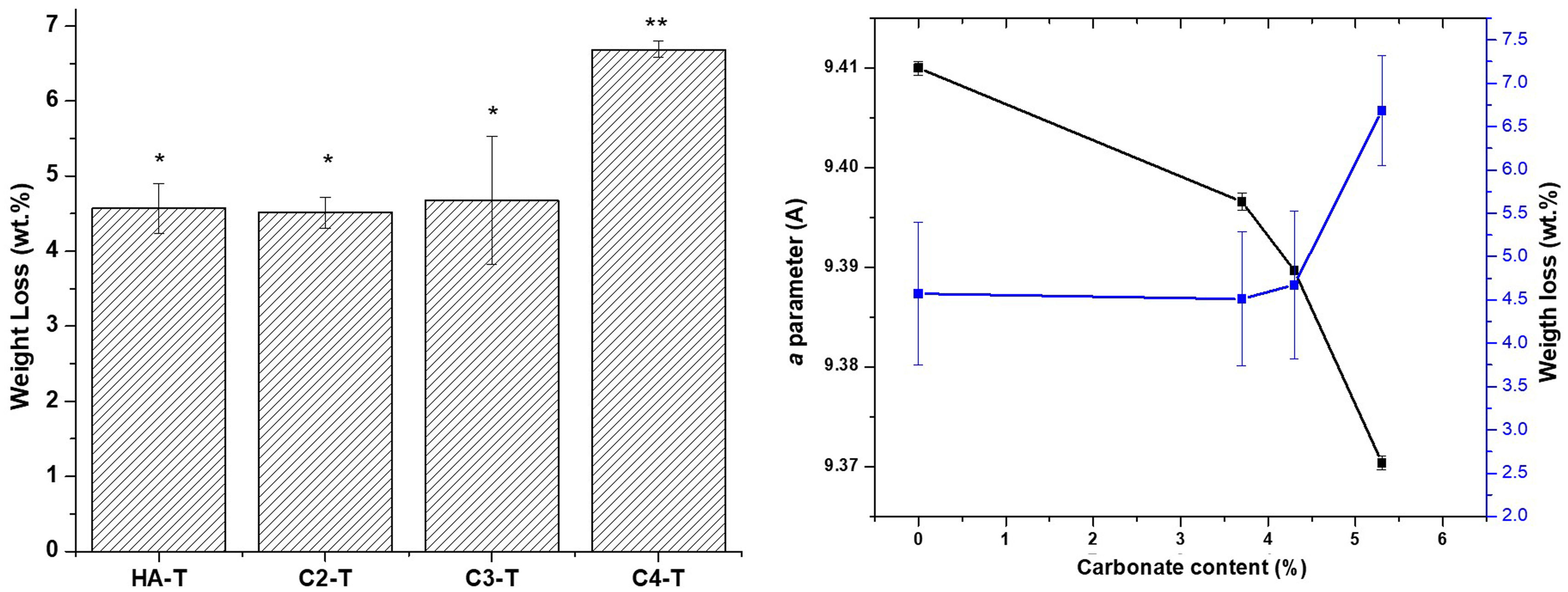
As in the case of strength, the similarities between the open porosities of the studied materials assure that the results on the effect of carbonate content on material degradation results would not to be masked by microstructural differences. Results of the accelerated degradation of the sintered materials in acidic conditions are presented in Fig. 8. There are no significant differences between the weight losses determined for the unsubstitued material (HA-T) and those corresponding to the substituted materials with the lowest carbonate contents (C2-T and C3-T). Conversely, the weight loss of the material with the highest carbonate content (C4-T) is significantly higher (>30wt.%) than those of the other three materials. In order to analyze this result, it is necessary to consider the weight loss associated to both dissolving ions, CO32− and PO43−, as discussed below.
. (a) Weight loss after 8h in acidic medium, groups identified by the same superscripts are not statistically different (p>0.05), (b) weight loss, carbonate content and the “a” parameter relationship.")
The accelerated dissolution process takes place by protonation of the CO32− to form carbonic acid and protonation of the PO43− to form phosphoric acid [Eqs. (1) and (2)] [60,61].
Therefore, the mass loss experienced by the materials is the sum of masses of the released phosphate groups, with higher molecular weight, carbonate groups and calcium ions.
The C2-T and C3-T would show weight losses like those of the HA-T because of the competition between the releases of the phosphate and the carbonate groups, the higher mass loss associated to the phosphate groups would mask that of the carbonate ones. For the material with the highest carbonate amount, the total mass loss due to ion release is higher because of a higher amount of carbonate being released towards the acidic solution.
The level of the weight loss for material C4-T (6.3±0.6wt.%) is similar to those reported for cementitious bone grafts. For example, Diez-Escudero et al. [40] reported weight losses of 5–8wt.% for a non-foamed carbonated calcium hydroxyapatite cement with higher content of carbonate (>11wt.%) and similar open porosity (≈60%) tested under the same conditions (solution, immersing time and specimen number) used in this work. This behaviour allows inferring that the CHA developed in this work would have osseointegration capability as the above-mentioned cements [60].
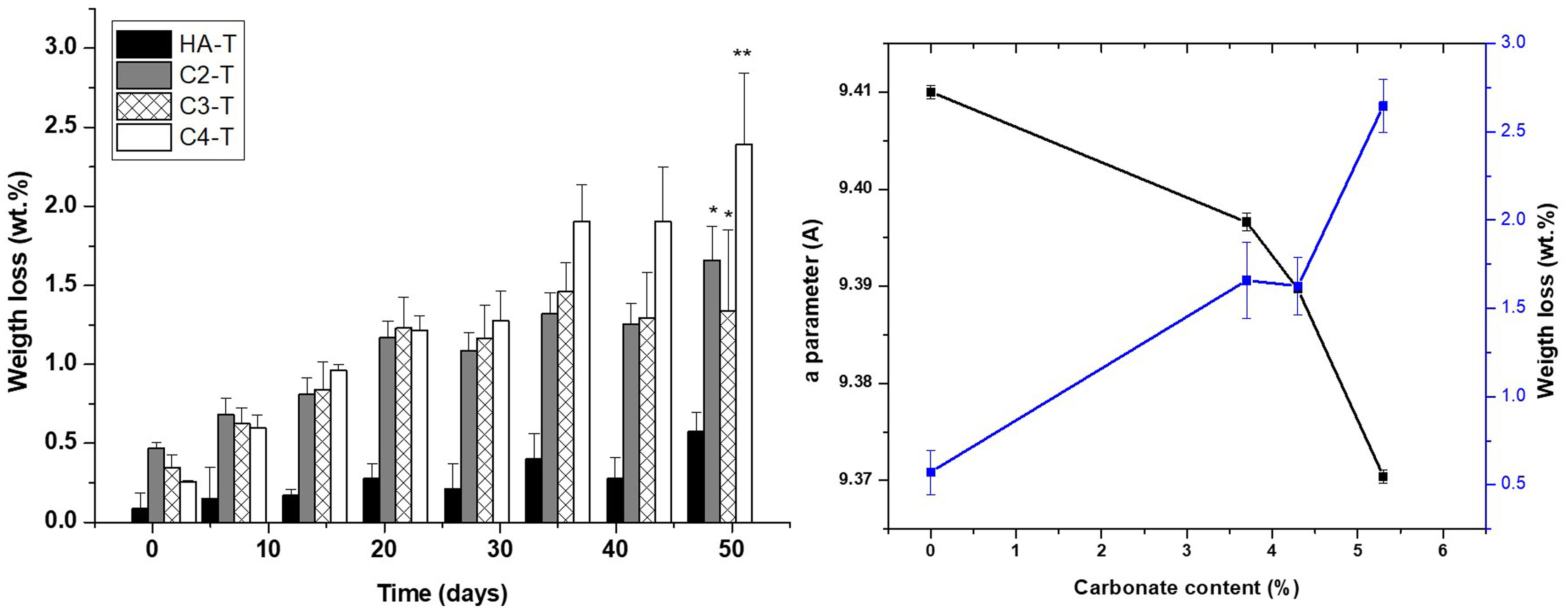
Results of the long-term degradation test Tris–HCl are plotted in Fig. 9. Unlike results on accelerated degradation, the weight losses of all substituted materials are higher than those of the unsubstitued HA-T for all time periods as expected from the chemical stability of stoichiometric HA in physiological environments [62]. Furthermore, there are no clear differences between the behaviours of the three substituted materials for the initial stages, up to 30 days immersion period, however, for longer times, the weight losses of the material with the highest carbonate content (C4-T) are significantly higher than those of C2-T and C3-T. The total mass loss due to ion release for the material with the highest carbonate amount is the largest when sufficient time for the release of the lower mass CO32− ion is given.
. (a) Weight loss after 7 weeks in physiological ambient, groups identified by the same superscripts are not statistically different (p>0.05), (b) weight loss, carbonate content and the “a” parameter relationship.")
Increased degradation of CHA sintered specimens in both tested environments, which simulate the physiological one, as compared to stoichiometric HA is due to the distortion in the lattice originated by CO32− substitution [63]. This fact is highlighted in Figs. 8 and 9(b), where the weight loss is related to the carbonate content and the “a” parameter of each material. This effect is the cause of the beneficial action of natural hydroxyapatite in the dynamic regeneration of bones. In the case of synthetic materials for bone regeneration, as considered here, the main problem of stoichiometric HA is the lack of capability of dissolution in such environment, which implies that HA implants remain in bones after years of implantation [64].
ConclusionsThree B-type carbonated hydroxyapatite (CHA) powders with 4–6wt.% were synthesized via the inverse route from stoichiometric hydroxyapatite and characterized.
The inclusion of carbonate ion causes a distortion of the crystalline lattice parameter of hydroxyapatite a and, in a lesser extent, of c. Powder morphology and thermal behaviour are modified according the carbonate level.
Sintered materials with similar microstructures, in particular, porosity levels, and three different carbonate amounts, slightly lower than those of the powders, have been processed from the HAC powders. The similarity between the microstructures has allowed to determine the sole effect of carbonate substitution on mechanical behaviour and bioactivity.
The carbonate substitution provides an increase in strength, higher for higher carbonate amounts. In vitro degradation rates in physiological and acidic environments are also improved, increasing with carbonate substitution.
The performance of the developed CAH materials, in particular the one of the highest carbonate ion substitution, allows to infer their adequacy to be considered for bone regeneration.
Support from Spanish National Research Council (CSIC) through the scientific cooperation project I – COOP+2018 (COOPA20289) and PIE 201660E097.
Support from Colombian Ministry of Science, Technology, and Innovation (MinCiencias) through project “111574557862” of 745/2016.

