





Infective endocarditis (IE) remains a severe and potentially fatal disease demanding sophisticated diagnostic strategies for detection of the causative microorganisms. The aim of the present study was to develop a broad-range 16S ribosomal RNA gene polymerase chain reaction in the routine diagnostic of IE for the early diagnosis of fatal disease. A broad-range PCR technique was selected and evaluated in terms of its efficiency in the diagnosis of endocarditis using 19 heart valves from patients undergoing cardiovascular surgeries at the Habib Bourguiba Hospital of Sfax, Tunisia, on the grounds of suspected IE. The results demonstrated the efficiency of this technique particularly in cases involving a limited number of bacteria since it helped to increase detection sensitivity. The technique proved to be efficient, particularly, in the bacteriological diagnosis of IE in contexts involving negative results from conventional culture methods and other contexts involving bacterial species that were not amenable to identification by phenotypic investigations. Indeed, the sequencing of the partial 16S ribosomal RNA gene revealed the presence of Bartonella henselae, Enterobacter sp., and Streptococcus pyogenes in three heart valves with the negative culture. It should be noted that the results obtained from the polymerase chain reaction-sequencing identification applied to the heart valve and the strain isolated from the same tissue were not consistent with the ones found by the conventional microbiological methods in the case of IE caused by Gemella morbillorum. In fact, the results from the molecular identification revealed the presence of Lactobacillus jensenii. Overall, the results have revealed that the proposed method is sensitive, reliable and might open promising opportunities for the early diagnosis of IE.
Conventional bacteriological diagnostic methods consist in the isolation and identification of the causative agent of an infection from a pathological product. This identification is based on the study of the phenotypic traits (morphological, cultural and biochemical) of the isolated bacteria. The microbiological diagnosis of infective endocarditis (IE) is mainly based on blood culture, excised cardiac valve tissue, or infected emboli. The conventional approach for the microbiological diagnosis of IE is successful in 92–95% of cases wherein a microorganism is present. Streptococci and Enterococci account for 45–60% of the cases of IE. Staphylococci represent the second most frequent group associated with IE. Various fungi or bacteria, such as Enterobacteriaceae, cause the remaining cases of IE. In fact, some bacteria are more specifically associated with IE, including those from the HACEK group (Haemophilus, Aggregatibacter, Cardiobacterium, Eikenella and Kingella).1
Although conventional cultures are negative in 5–8% of IE cases, IE is a challenging disease to diagnose. This is particularly due to the wide variation in the criteria used to define it even after surgery. Cultures and microscopy may not reveal the causative organism in the infected valve. In such cases, slow-growing or non-culturable microbes may be the etiologic agents, and the patient may have received antimicrobial treatment at the time the specimen was obtained.2 In addition, phenotypic identification techniques are not applicable for bacteria expressing little discriminating phenotypic characteristics. In recent years, researchers have increasingly become interested in the search for viable strategies and techniques to overcome the inadequacies and limitations associated with the conventional microbiological methods of IE diagnosis.
The literature indicates that culture-independent molecular techniques that involve the use of broad-range PCR to detect and amplify bacterial DNA and sequencing of the genes coding for 16S rRNA may open promising opportunities for establishing the etiology of the infection. In fact, the use of these techniques has revolutionized the detection and analysis of infectious diseases and the ways to combat them.2,3 Furthermore, the literature indicates that the nested-PCR approach offers a rapid, efficient and reliable tool for the detection of bacterial infections from clinical specimens.4,5 Some studies have, however, reported that the frequent incidence of false-positive results is a major problem hampering the application of nested PCRs in large-scale clinical settings.4,6 Accordingly, the literature highlights the importance of the amplification of part of the 16S rRNA gene and direct sequencing of the amplicon to identify microbes in valve tissues. The determined sequence is often compared to the reference sequences, a procedure that often allows for the identification of the causative agent. The results of conventional bacteriological or/and histopathological diagnostic methods for IE have often been compared to those obtained by PCR.1,2 The broad-range PCR appeared a relatively easy and reliable method that gives accurate results when applied to surgically removed heart valves of patients with IE and may be used as an adjunct for cases where cultural methods fail.2 The amplification of the gene that encodes the 16S rRNA of eubacterium-specific sequences from heart valve tissues seem to offer a new promising tool for the etiological diagnosis of IE. This method has allowed for the detection of fastidious organisms, such as Tropheryma whipplei, Coxiella burnetii and Bartonella spp, which require special culture conditions.1,2 The broad-range PCR technique has also proved efficient for confirming the presence of several microorganisms responsible for IE, including Granulicatella elegans, Streptococci, Staphylococci, Enterobacter, Borrelia burgdorferi, Candida albicans, and Aspergillus species.7,8 Lactobacilli have been identified in some clinical reports as causal agents of IE, meningitis and several other health diseases.9
Considering the growing global concerns over the prevalence of IE and the promising potential that molecular techniques might open for the identification and analysis of this serious infection, the present study aimed to investigate the feasibility and potential of a broad-range PCR technique, based on bacterial 16S rRNA gene, in the diagnosis of IE.
Materials and methodsBacterial strains and specimensThe reference strains used as control organisms in the PCR performed in this study were Escherichia coli ATCC 25922, Staphylococcus aureus ATCC 43300, Streptococcus pneumoniae ATCC 49619, Haemophilus influenzae ATCC 1399, Salmonella Typhimurium WHO 32, Klebsiella pneumoniae WHO 1, Enterococcus faecalis ATCC 159, Citrobacter freundii WHO 9, Pseudomonas aeruginosa WHO 8, and Serratia marcescens WHO 10. Some other strains that posed identification problems were also isolated from the heart valves of the patients, namely Acinetobacter baumannii, Micrococcus lylae, and Gemella morbillorum. The isolates of each of the above bacteria were obtained from the laboratory of clinical microbiology in the Habib Bourguiba hospital of Sfax, Tunisia.
Suspensions of E. coli ATCC 25922, S. aureus ATCC 43300, and S. pneumoniae ATCC 49619 containing (106, 105, 104, 103, 102 and 10CFU/mL) were also prepared by routine culture and dilution counts.10
The study used a total of 19 heart valves from patients who underwent heart valve replacement surgeries (Prosthetic valve) at the cardiovascular service of the Habib Bourguiba hospital of Sfax, Tunisia, on the grounds of suspected IE.
DNA extractionThe DNA of the bacterial strains of each specimen valve tissue was extracted by the Cetyltrimethylammonium Bromide–Phenol-Chloroform/Isoamyl Alcohol method ((CTAB)–Phenol-Chloroform/Isoamyl Alcohol). Prior to (CTAB)–Phenol-Chloroform/Isoamyl Alcohol extraction, 500μL of lysis buffer (200mM NaCl, 20mM Tris HCl, pH 8, 50mM EDTA, pH 8, and 1% SDS) and 25μL of Proteinase K (10mg/mL) (Sigma) were added to approximately 10mg of valve tissue. The mixture was then vigorously agitated and incubated at 65°C for 30min or until the complete dissociation of valve tissue fragments. The enzymatic reaction was stopped by incubation at 95°C for 10min, and samples were centrifuged at 10000×g for 5s. The samples were then harvested by centrifugation at 14000×g for 20min.
After homogenization, the samples were incubated in a solution of (CTAB)-NaCl (100μL of 5M NaCl and 80μL of 10% CTAB) for 10min at 65°C, and then mixed with 750μL of chloroform–isoamyl alcohol (24:1 [vol/vol]) and centrifuged for 15min at 14000×g in an Eppendorf centrifuge. The aqueous phase was separated, mixed with 750μL of phenol chloroform–isoamyl alcohol (25:24:1 [vol/vol/vol]), and centrifuged for 15min at 14000×g in an Eppendorf centrifuge. The obtained aqueous phase was reseparated and mixed with an equal volume of isopropanol. The samples were left to rest for 1h at 80°C and then centrifuged for 15min at 14000×g. The DNA pellet was washed with 70% ethanol, air-dried, and dissolved in a final volume of 100μL of TE buffer. The DNA extraction for each bacterial dilution was performed using (CTAB)–Phenol-Chloroform/Isoamyl Alcohol extraction and commercial kits (QIAamp® DNA Mini Kit) in accordance with the manufacturers’instructions. To compare the relative efficacy of the both extraction methods.
The genomic DNA of the isolates was extracted with the traditional boiling method as described by Harris et al. (2003). In brief, 6 bacterial colonies was re-suspended in 1mL of sterile saline and boiled for 10.
DNA concentration was estimated by gel migration.
Pretreatment of TaqDNA polymerase to remove contaminating bacterial DNAPrior to the PCR amplification, a pretreatment protocol was developed to prevent the occurrence of false positives due to the presence of Thermophilus aquaticus DNA in the Taq polymerase. In brief, the water, buffer, deoxynucleoside triphosphates, and Taq DNA polymerase components were mixed and incubated for 60min at 37°C with 1U, 1.5U and 2U of DNase I (Amersham Biosciences). The enzyme was then inactivated by incubation at 90°C for 30min, followed by the addition of primers and template DNA. The PCR amplification phase was then started. The different components were also mixed and incubated with 1U of the restriction enzymes Sau 3AI and AluI.4,6
Broad-range PCR amplification of 16S rRNA genes and sequencingInitially, the extracted DNA was tested to check the absence of PCR inhibitors in the valve tissue by the amplification of 209bp fragment of a human β-globin gene. β-Globin positive samples were then examined by broad-range PCR.11
For the nested PCR, primers described by Wilkinson et al.5 were used in the present study.
The first-round PCR reaction was carried out in a reaction mixture of 50μL per tube containing 10μL of l0× PCR buffer, 0.4μM of each PCR primers (NW9/NW17, NW9/NW12, NW11/NW17 or NW11/NW12), 0.2μM of deoxynucleoside triphosphates, and 0.0025U of Taq polymerase. Two oligonucleotide primer pairs, NW9 (5′-GCTAACTAACGTGCCAGCAG-3′) and NW17 (5′-TAAGGGCCATGA (T/G)GA(C/T)TTGAC-3′), were used. PCR was performed using the Gene-Amp PCR System 9700 (Perkin Elmer Cetus). The PCR protocol consisted of an initial cycle at 95°C for 5min, followed by 40 cycles of denaturation at 94°C for 30s, annealing at 50°C for 30s, and extension at 72°C for 45s, with a final cycle at 72°C for 7min. Each PCR run included a positive control (E. coli) and 2 negative controls (previous negatively tested samples and distilled water). The second-round PCR was carried out with the same volume and reagents mentioned above using primers NW11 (5′-GAGGCCTACAAGCGGTGGAG(C/G)ATGTG-3′) and NW12 (5′-GAGGCCTCCAACATCTCACGACACGAG-3′). The thermocycling parameters were identical to the ones used in the first round, except for the annealing temperature, which was increased to 58°C, and extension step, which was performed at 72°C for 30s. An amount of 5μL of DNA extract was added in the first-round PCR, and 1μL of the PCR product was added for the reamplification round. The purified PCR products were sequenced with an ABI PRISM BigDye sequencing kit and analyzed with an ABI 3100-Avant genetic analyzer (Applied Biosystems).
Phylogenetic analysisThe program Molecular Evolutionary Genetic Analysis software, ver. 7.0 (MEGA7.0; http://www.megasoftware.net) was performed in order to edit and align the sequence files, which were manually adjusted. In this study, phylogenetic tree was generated using maximum parsimony (MP) in MEGA5.0. Bootstrap values for the maximum parsimony tree (MPT) were caculated for 1000 replicates. The edited fragment (155bp) of the 16S rRNA sequences were compared with other available Lactobacillus species sequences in the GenBank. Furthermore, the sequences of some known species of Lactobacillus were downloaded from GenBank and used to reconstruct 16S rRNA region phylogenetic trees.
PCR-RFLPThe PCR products from cultures and clinical specimens were digested using 10–17μL of PCR amplified DNA restricted with 1U of EcoR I, Fok I and DdeI in a total volume of 20μL according to the manufacturer's instructions (Invitrogen) and incubated for 4h at 37°C. The restriction fragments were separated by electrophoresis on agarose gel 3%.
The search for recognition sites of restriction enzymes at the nucleic acid sequences was performed using the REBASE program (http://rebase.neb.com/rebase/rebase.html). This program has commonly been used for the identification of predicted enzymes and estimation of the size of digested amplicon fragments (Genbank sequences).
Results and discussionComparison of DNA extraction methodsTwo different DNA extraction methods were examined to ascertain their relative effectiveness for extracting bacterial DNA from gram positive and negative bacteria. The CTAB method was the most effective for the detection of DNA prepared from the pure bacterial culture of Gram-positive and negative bacteria (positivity was observed till the suspension 102CFU/mL for S. pneumoniae (Fig. 1A) and 10CFU/mL for S. aureus and E. coli). Although the kit method was less effective in the extraction of bacterial DNA, positivity was observed till the suspension 104CFU/mL for all the tested strains.
 First-round PCR amplification with primer pairs MW9/NW17. Lane 1 DNA ladder; lane 2–7 contained different suspensions of Streptococcus pneumoniae: lane 2 – 106CFU/mL; lane 3 – 105CFU/mL; lane 4 – 104CFU/mL; lane 5 – 103CFU/mL; lane 6 – 102CFU/mL; lane 7 – 10CFU/mL; lane 8 – negative control. (B) Second-round of PCR amplification with primer pairs NW11/NW12. Lane 1 DNA ladder; lane 2, 3 contained 1μL from the first-round PCR; lane 2 – suspension 102CFU/mL of Streptococcus pneumoniae; lane 3 – suspension 10CFU/mL of Streptococcus pneumoniae; lane 4 contained 1μL of the negative reaction mixture from the first round. (C) Second-round of PCR amplification of the negative reaction mixture with primer pairs NW11/NW12. Lane 1 DNA ladder; lane 2–6 contained 1μL of the negative reaction mixture from the first round; lane 2 negative reaction mixture without pretreatment; lane 3–6 negative reaction mixture treated with 1U, 1.25U, 1.5U and 2U of DNase I, respectively.")
Nested-PCR using the primer pairs MW9/NW17 and NW11/NW12. (A) First-round PCR amplification with primer pairs MW9/NW17. Lane 1 DNA ladder; lane 2–7 contained different suspensions of Streptococcus pneumoniae: lane 2 – 106CFU/mL; lane 3 – 105CFU/mL; lane 4 – 104CFU/mL; lane 5 – 103CFU/mL; lane 6 – 102CFU/mL; lane 7 – 10CFU/mL; lane 8 – negative control. (B) Second-round of PCR amplification with primer pairs NW11/NW12. Lane 1 DNA ladder; lane 2, 3 contained 1μL from the first-round PCR; lane 2 – suspension 102CFU/mL of Streptococcus pneumoniae; lane 3 – suspension 10CFU/mL of Streptococcus pneumoniae; lane 4 contained 1μL of the negative reaction mixture from the first round. (C) Second-round of PCR amplification of the negative reaction mixture with primer pairs NW11/NW12. Lane 1 DNA ladder; lane 2–6 contained 1μL of the negative reaction mixture from the first round; lane 2 negative reaction mixture without pretreatment; lane 3–6 negative reaction mixture treated with 1U, 1.25U, 1.5U and 2U of DNase I, respectively.
The literature indicates that the extraction of bacterial DNA could be accomplished with different degrees of efficiency, depending on the method used.13,14 In the present study, the application of the in-house sample preparation methods showed higher levels of efficiency. In fact, the case involving manipulation from complex matrices, such as pathological products, suggested that the samples could presumably contain various inhibitors that need to be eliminated before the PCR.15 Accordingly, several studies, have previously reported on the extraction efficiency of CTAB, and its promising potential for the enhancement of PCR sensitivity.11,16,17
Validaton of the broad-range PCR on the reference strainsThe introduction of the broad-range PCR in the laboratory of clinical bacteriology requires its validation on reference strains before its application on bacterial strains of uncertain identification or pathological products.12 To ensure the specificity of the broad-range PCR, the PCR products of 10 reference strains were sequenced using the primers NW11 (fragment length of 263bp) and NW12 (fragment length of 584bp). The determination of these two sequences allowed for the detection of the sequence of a region of 155bp. The sequences of the three different fragments (263bp, 584bp, 155bp) of the 16S rRNA gene of each reference strain were compared using the BLAST program. The results revealed that the reference strain under investigation showed high levels of sequence identity. The results obtained for the sequence data on the molecular identification of bacteria of these strains using a small fragment (155bp) of the 16S rRNA gene confirmed the discriminating potential of the broad-range PCR technique.
Application of nested PCR techniqueThe efficiency of the nested PCR technique was evaluated using three bacterial suspensions of the three reference strains (S. aureus, S. pneumoniae and E. coli) containing a decreasing number of bacteria (106, 105, 104, 103, 102 and 10CFU/mL). The amplification of these extracted DNA using the universal primers NW9/NW17 during the first round of PCR allowed for the amplification of the 16S rRNA gene of a bacterial suspension containing ≥102CFU/mL (Fig. 1A). The second PCR using the universal primers NW11/NW12 was applied to a bacterial suspension containing a number of bacteria ≤102CFU/mL. The 16S rRNAs of 155bp were amplified in the expected size and with high intensities for the different bacterial suspensions tested (Fig. 1B). However, the results showed the presence of an amplification with the same size of the positive control and with high intensity for all the negative controls after the second-round of PCR (Fig. 1B). No amplification of this type was observed in the first-round PCR. A similar result was previously reported by Carroll et al.6 who detected amplification in the negative control of the reaction after the nested PCR.
The technique of the nested PCR targeting the16S rRNA gene using universal and species-specific primers has been performed in several studies.3–5,12 The technique has also been used in various cases of low abundance organisms to increase sensitivity.5,6,13 The two-step process of the nested PCR technique resulted in the dilution of potential inhibitors and provision of sufficient template for the secondary reaction. Almost all the samples that gave negative results in the primary PCR yielded into a specific product after nested PCR, indicating the presence of bacterial DNA.13
However, the present results suggest that despite the improved sensitivity achieved by the application of the nested PCR, the second amplification could increase the risk of false positive occurrences. Many studies have reported that unlike the case with species-specific PCR, the validity of broad-range PCR techniques for the amplification of the 16S rRNA gene of broad spectrum of pathogenic bacteria is compromised by the high rate of false positive results.18,19 This method is more vulnerable to contamination than species-specific PCR.20 In the nested PCR, the second-round PCR performed on the amplification product from a previous PCR may involve contamination, thus increasing the rate of false positives.4,6 Such DNA contamination can occur due to improper manipulations (personnel, equipment, reagents, etc.) or during various experimental phases (extracting DNA samples or preparation of PCR reaction mixture).5,14,21 In the present study, all necessary precautions were taken to avoid the emergence of potential contamination due to handling or operational mistakes. The different steps of bacterial DNA extraction, reaction mixture preparation, and amplification execution were performed in separate rooms as previously described elsewhere.20–22 Furthermore, all reagents and equipments used in the PCR experiments were regularly decontaminated before use (pipettes, tips, sterile water, buffer, etc.). The preparation of the reaction mixture and deposition of the extracted DNA were carried out in a laminar flow hood under sterile areas. Nevertheless, and despite all these precautions, an incidence of contamination from exogenous bacterial DNA was observed during the second-round of the nested PCR. In fact, numerous studies have reported that the Taq polymerase is the major source of contamination with exogenous bacterial DNA (Hughes et al., 1994; Rand and Houck, 1990).23 The literature presents several methods of contaminated DNA removal to avoid this problem.6,18,19 In addition to the proper laboratory organization and adequate separation of the different PCR stages21 the techniques proposed for the elimination of contamination by exogenous bacterial DNA include the UV irradiation of the different gradients in the reaction mixture, and the pretreatment of the reaction mixture by restriction enzymes or DNase I.6,18,23 The long UV exposure technique has, however, been reported to reduce the activity of Taq polymerase.23
Pretreatment with restriction enzymes and with DNase IThe results recorded after the treatment of the reaction mixture with a high concentration of the two restriction enzymes (Alu I and Sau 3AI) revealed the absence of amplification for all samples (negative and positive controls). This could be attributed to the presence of a high salt concentration by the uploading buffer of the restriction enzyme, which, in turn, could have induced the inhibition of the Taq polymerase. Many studies have reported that the pretreatment of the reaction mixture before the addition of DNA template with low concentrations of restriction enzymes failed to eliminate contamination. This failure could be ascribed to the absence of cleavage sites for both restriction enzymes at the exogenous DNA due to the unknown source of the contaminating DNA, since the Taq polymerase may contain DNA of unknown origin.23,24
The negative reaction mixture was pretreated with different concentrations of DNase I and exposed to various incubation periods during the first and second-round of the PCR. This pretreatment allowed for the elimination of contamination caused by exogenous bacterial DNA. The absence of amplification in the negative control (water and reaction mixture) was observed after the pretreatment with DNase I at a concentration of 1.5U and after an incubation period of 60min at 37°C and 30min at 90°C. The DNase I pretreatment process was developed by Heininger et al.18 who assayed DNase I at different concentrations and reported on its efficiency in the elimination of false positive results. The concentration required for DNase I to remove the exogenous DNA of the Taq polymerase depended on the nature of the Taq polymerase (Go-Taq polymerase, Deep Vent Exo-polymerase, super-Taq polymerase, Ampli-Taq polymerase, etc.) and the level of enzyme contamination.18,19 The cloning and sequencing of the PCR fragment produced by Promega Taq polymerase revealed the presence of at least 5 different clones, confirming that the Taq polymerase contained the unknown DNA from more than one bacterial species.25
The nested PCR was applied (the primer pairs NW9/NW17 for the first-round PCR, and NW11/NW12 for the second-round PCR) after the pretreatment of the reaction mixture with 1.5U of DNase I (60min at 37°C and for 30min at 90°C) and by adding different bacterial suspensions of S. pneumoniae (106, 105, 104, 103, 102 and 10CFU/mL). The results revealed PCR positivity only for the suspensions containing a number of bacteria ≥103CFU/mL. Thus, the results obtained by nested PCR after reaction mixture pretreatment with DNase I at the first and second-rounds of PCR revealed that the PCR efficiency decreased when compared to the one observed for the broad-range PCR using the primer NW9/NW17 without reaction mixture pretreatment with DNase I. In fact, the broad-range PCR allowed for the amplification of bacterial suspensions of S. aureus and E. coli containing 10CFU/mL. The present study has shown that the pretreatment of the reaction mixture by DNase I may damage the activity of the Taq polymerase and, therefore, induce the occurrence of false negative results. Thus, despite offering the advantage of eliminating contaminated DNA in Taq DNA polymerase, the pretreatment of the reaction mixture by the DNase I may reduce PCR sensitivity. The decrease in PCR sensitivity could presumably be attributed to the exposure of Taq DNA polymerase to elevated temperatures for a long period of time.23 Accordingly, the optimization of DNase I treatment efficiency requires the acquisition of the most durable taq polymerase available.23 Overall, and in spite of its limited ability to fully denature the enzyme without compromising Taq polymerase activity, DNase I pretreatment seems to be the most effective contamination removal method.23
Effect of amplicon size on broad-range PCR sensitivityTo investigate the potential enhancement of PCR sensitivity, DNA amplification by broad-range PCR was further assayed using 4 primer pairs, namely NW9/NW17 (amplify a region of 710bp), NW9/NW12 (amplify a region of 600bp), NW11/NW17 (amplify a region of 280bp), and NW11/NW12 (amplify a region of 155bp). The amplification of the DNA extract from the reference S. pneumoniae strain using these 4 primer pairs revealed amplification in the expected size. The intensity of the PCR fragment was noted to increase with the decrease of amplicon size. The most intense band was observed with the NW11/NW12 primer pairs, which amplified a region of 155bp. In fact, the shorter the bacterial studied fragments were, the higher the possibility of bacterial identification in cases involving a small number of bacteria became.
The first study to use broad-range PCR targeting the full 16S rRNA gene for the detection of bacterial DNA in synovial tissues used automated DNA extraction techniques, which differs from the in-house sample preparation method used in the present study.26 In fact, the T4 Gene 32 Protein should also be added to the PCR mix. Although the last technique proved efficient, its application is costly, time-consuming and high-expertise requiring. The diagnostic application of the broad-range amplification–sequencing method requires adequate databases with good quality and full-length sequences of clinically important bacteria to allow genus or species identification of bacterial pathogens, either directly from clinical specimens or from cultures.2 Although long amplicons can provide more information, they are more difficult to generate, which results in the loss of sensitivity.3 Longer targets (e.g., primers 27f (forward)/1492r (reverse)) may be used when bacteria are abundant in the samples.28 However, Schuurman et al.20 used the primer pairs that amplified a 1500bp fragment of 16S r RNA gene and another step of reamplification to increase the sensitivity of the technique. The identification by broad-range PCR amplification and partial sequencing of the 16S rRNA gene directly from clinical specimen and culture was previously reported in several studies proving the efficiency of the method to diagnose IE.1,2,22,27,29
Contribution of the broad-range PCR to the diagnosis of IEThe present study examined 19 heart valves from patients who underwent valve replacement surgeries at the cardiovascular service of the Habib Bourguiba hospital of Sfax, Tunisia, on the grounds of suspected IE. The conventional bacteriological examination of the heart valves showed the presence of 3 positive cases: one case of A. baumannii, a case of G. morbillorum, and another case of M. lylae. DNA extraction was followed by the amplification of a 209bp fragment of the human gene encoding β-globin. The results showed the absence of amplification except for one heart valve. This could be explained by the presence of PCR inhibitors. The amplification of the gene for human β-globin was used as a control to verify the absence of PCR inhibitors in the DNA extracts from clinical samples.1,21
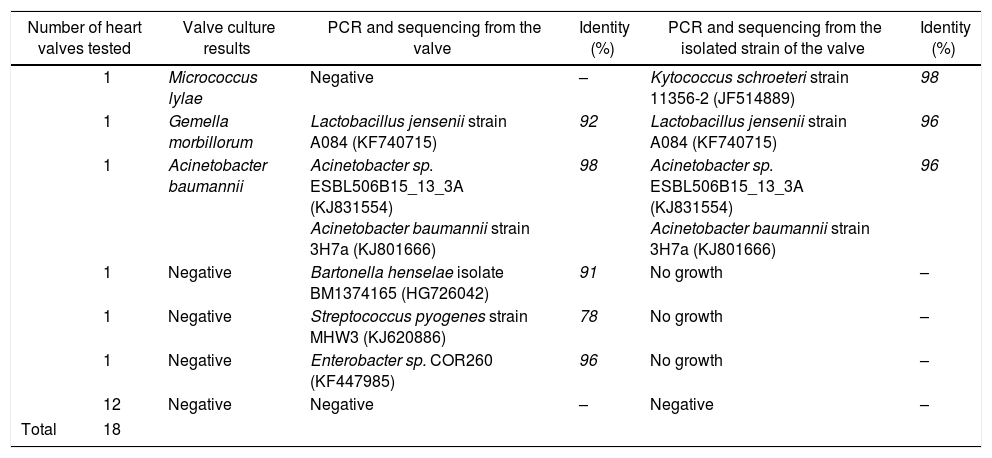
To investigate the contribution of the broad-range PCR for the diagnosis of IE, the bacterial 16S rRNA gene was amplified using primer pairs NW11/NW12 from 18 heart valves that had previously shown the absence of PCR inhibitors. The results revealed amplifications in the expected sizes for 5 heart valves. The comparison between the results obtained by PCR to those from the culture valves revealed that the broad-range PCR showed a higher level of positivity (Table 1). Moreover, three heart valves that had been found negative by conventional bacteriological examination were labeled positive by broad-range PCR. Two cases were considered positive by both conventional microbiological method and broad-range PCR technique. The results also revealed a unique instance wherein a heart valve was labeled as negative by the broad-range PCR but positive by the culture and β-globin PCR techniques. Our result was similar to those reported by Gauduchon et al.1 and Vondracek et al.29 The negative PCR result in this case may have been due to three factors: the long interval between antibiotic treatment and surgery; the use of an inappropriate valve fragment for PCR (the actual infected site of the heart valve was not analyzed) or the presence of PCR inhibitors (although extraction and amplification controls gave the expected results, and amplification of the beta-globin gene was positive).1,29
Results of broad-range bacterial PCR for heart valves and strain isolated from the same tissue.
| Number of heart valves tested | Valve culture results | PCR and sequencing from the valve | Identity (%) | PCR and sequencing from the isolated strain of the valve | Identity (%) | |
|---|---|---|---|---|---|---|
| 1 | Micrococcus lylae | Negative | – | Kytococcus schroeteri strain 11356-2 (JF514889) | 98 | |
| 1 | Gemella morbillorum | Lactobacillus jensenii strain A084 (KF740715) | 92 | Lactobacillus jensenii strain A084 (KF740715) | 96 | |
| 1 | Acinetobacter baumannii | Acinetobacter sp. ESBL506B15_13_3A (KJ831554) Acinetobacter baumannii strain 3H7a (KJ801666) | 98 | Acinetobacter sp. ESBL506B15_13_3A (KJ831554) Acinetobacter baumannii strain 3H7a (KJ801666) | 96 | |
| 1 | Negative | Bartonella henselae isolate BM1374165 (HG726042) | 91 | No growth | – | |
| 1 | Negative | Streptococcus pyogenes strain MHW3 (KJ620886) | 78 | No growth | – | |
| 1 | Negative | Enterobacter sp. COR260 (KF447985) | 96 | No growth | – | |
| 12 | Negative | Negative | – | Negative | – | |
| Total | 18 | |||||
The identification of the bacteria detected in the five positive heart valves by broad-range PCR was performed after sequencing, using the sense primer NW11, and alignment, using the BLAST program (Basic Local Alignment Search Tool). The sequencing of the partial 16S rRNA gene revealed the presence of Bartonella henselae, Enterobacter sp, and Streptococcus pyogenes in three heart valves that were negative (Table 1). The negativity of culture in the case of Bartonella could be due to the difficulty of isolating the bacteria by conventional microbiological methods. Hadfield et al.30 reported on the first case of endocarditis caused by B. henselae using an approach similar to the one described in this study. Several studies have also used this technique to detect Bartonella in the aortic valve of patients with culture-negative IE.31,32 The finding of two heart valves to be culture negative but PCR positive in the cases of Streptococcus and Enterobacter was previously attributed to prior antimicrobial treatment of patients.2,22 Various studies have also reported cases of endocarditis due to Streptococcus and Enterobacter.8,33
The PCR based methods are rapid, reliable and highly sensitive tools for the microbiological diagnosis of culture negative IE. This method can substantially improve the diagnostic outcome of microbiological examination of excised heart valves not only for fastidious, slow growing, and/or non-culturable microorganisms but also for easy-to-culture pathogens, such as streptococci and staphylococci.1,8,27 The described molecular approach proved to be superior to bacterial culture.1,2,22,27,29
In the present study, a good concordance was observed between PCR and conventional bacteriological diagnosis in the case of IE caused by A. baumannii. Ryczak et al.34 reported that this bacterium was responsible for IE. The application of broad-range PCR and sequencing showed an agreement between the sequence amplified from the DNA extracted from the strain isolated and the one extracted from the tissue from the same heart valve. However, the results obtained from the PCR sequencing identification from the DNA extracted, from a heart valve was not consistent with the one found by the conventional microbiological methods in the case of IE caused by G. morbillorum. In fact, the molecular identification techniques revealed the presence of Lactobacillus jensenii (Table 1), which has also been described by many studies.35,36 To confirm the results from PCR applied to the DNA extracted from the heart valve, the present study opted for the identification of the strain isolated from the same valve. The sequencing of the amplified 16S rRNAgene from the isolated strain (G. morbillorum) indicated (96% similarity to L. jensenii) that the strain was L. jensenii. A tree was constructed by the neighbor-joining method based on extracted DNA from isolated strain and the one from the heart valve, showing the phylogenetic position of the strain and the valve related to L. jensenii (Fig. 2). The phylogenetic tree analysis clearly demonstrated that the 16S rRNA of the strain and the valve were localized within the L. jensenii clade.

This broad-range PCR, therefore, represents a highly accurate and versatile method for bacterial identification even when the species in question are notoriously difficult to identify by phenotypic means. The literature has confirmed the importance of 16S rRNA amplification and sequencing as compared to the conventional microbiological methods in the identification of microorganisms with uncertain characterization by phenotypic tests.1,36,37 However, Woo and colleagues38,39 have proven that the identification of Lactobacillus and Gemella species by culture methods is insufficient and that the PCR of the 16S rRNA gene, followed by sequencing, is the method of choice for the identification of such microorganisms. Woo et al.39 have also reported that the species of Gemella are important causal agents of IE. For the patient with negative PCR and positive cultures, the strain isolated from valves was identified as M. lylae by conventional microbiological methods such as ID32 STAPH gallery. To confirm the bacterial identification, the analysis proceeded by amplifying and sequencing a part of the 16S rRNA gene of this strain. The PCR results indicated that the strain was Kytococcus schroeteri (Table 1). Our results were in accordance with what was previously reported by Mnif et al.40 They have reported that biochemical identification does not differentiate between M. lylae and K. schroeteri and that further molecular studies are needed to achieve more identification that is accurate. Becker et al.41,42 reported cases of endocarditis caused by K. schroeteri. These authors have confirmed that biochemical identification does not differentiate between M. lylae and K. schroeteri and that further molecular studies are needed to achieve more accurate identification.
The sequences of the 16S rRNA gene of the isolates identified in the present study exhibited about 97% of similarity to known sequences available database, which provided the eventual genus identification. Goldenberger et al.2 previously reported that, during their study, several reference sequences in the databases often presented only partial sequences, which is consistent with the results of the present study. However, these authors suggested that, in some cases, the 16S rRNA gene sequence analysis cannot be employed as the sole technique for species identification. Nested primers that span unique sequences in the 16S rRNA gene in staphylococci and streptococci at the species level (species-specific PCR) were used, and their application was justified by the need to identify the most commonly anticipated species.3,27 PCR-RFLP was developed for the detection and identification at the species level of Bartonella in clinical specimens.43
PCR-RFLPFurther tests based on the PCR-RFLP technique were performed to verify the specificity of the amplification and confirm the findings obtained by the identification via sequencing from the heart valve and the strain isolated from the same material. The REBASE program was used to search for recognition sites of restriction enzymes in the nucleic acid sequence. To confirm the case of the IE with L. jensenii, the PCR product of 710bp of the strain isolated and identified as G. morbillorum based on the culture was digested by EcoR I. This restriction enzyme cut one time the sequence of 710bp of the 16S rRNA gene of G. morbillorum and did not digest the same amplified region coming from L. jensenii. The obtained results did not show a restriction site in the amplified region, confirming that the detected bacterium was L. jensenii (Fig. 3A). Likewise, the use of Fok I and Dde I restriction enzymes which cut one time the PCR products of 155bp for L. jensenii, but not that of G. morbillorum, will provide further support that the detected strain was a L. jensenii and not G. morbillorum. In the present work, the PCR product of 155bp obtained from the heart valve of a patient operated for suspicion of IE with G. morbillorum was submitted to digestion with Fok I and Dde I. The results showed a restricted fragment in the expected sizes (Fig. 3B) of 105bp and 48bp after digestion with Fok I and two fragments of 76bp after digestion with Dde I.
 PCR product amplified from the isolated strain; lane 1 DNA ladder; lane 2 PCR product non-digested; lane 3 PCR product digested with the enzyme EcoRI. (B) PCR product amplified from the heart valve; lane 1 DNA ladder; lane 2 PCR product non-digested; lane 3 PCR product digested with the enzyme Fok I; lane 4 PCR product digested with the enzyme DdeI.")
PCR-RFLP from the heart valve and the strain isolated from patients with suspected infective endocarditis with the enzymes EcoRI, FokI and DdeI. (A) PCR product amplified from the isolated strain; lane 1 DNA ladder; lane 2 PCR product non-digested; lane 3 PCR product digested with the enzyme EcoRI. (B) PCR product amplified from the heart valve; lane 1 DNA ladder; lane 2 PCR product non-digested; lane 3 PCR product digested with the enzyme Fok I; lane 4 PCR product digested with the enzyme DdeI.
Further assays based on the PCR-RFLP technique were performed to confirm the case of IE with B. henselae identified after PCR and sequencing of tissues from valves with negative culture. The restriction enzyme Sau 3AI cut two folds in the region of 155bp of B. henselae. The digestion of the PCR product (155bp) with Sau 3AI showed three fragments in the expected size (66bp, 53bp, 39bp). Sau 3AI did not cut the region of 155bp of Bartonella quintana, thus establishing B. henselae as the eventual infecting species in this case. The implication of the Bartonella species in a variety of human diseases and the difficulty of its isolation from clinical specimens highlight the need for the development of better detection and identification methods.43 In fact, both B. quintana and B. henselae were found to cause endocarditis and previously identified by PCR-RFLP.32,43 This assay might open new promising opportunities for the early diagnosis of IE and accurate differentiation between species.43
ConclusionThe present study has confirmed that the broad-range amplification–sequencing method allowed for the achievement of a more sensitive clinical diagnosis of endocarditis. This PCR method proved to be superior to conventional methods in detecting bacteria from heart valves. The application of this molecular technique, especially in the case of negative results, substantially improved the diagnosis of not only highly fastidious and slow-growing microorganisms but also pathogens that are generally considered easy to cultivate. In addition, the broad-range PCR showed to be effective in the identification of microorganisms whose characterization by phenotypic tests was uncertain. The PCR technique proposed in this study also offers a simple, user-friendly, time-saving and cost-effective tool for the identification of IE. Thus, this technique may be a promising tool for application in the routine testing of infectious diseases.
Conflicts of interestThe authors confirm that there are not conflict of interest for this article.
The authors would like to express their sincere gratitude to Mr. Anouar Smaoui and Mrs. Hanen Ben Salem from the English Language Unit at the Faculty of Science of Sfax for their valuable and language polishing and editing services.