


Klebsiella pneumoniae is important human and animal pathogen that causes a wide spectrum of infections. In this study, isolates from cattle nasal swabs samples were identified by 16S rRNA, and to evaluate the antimicrobial susceptibility, virulence gene carrying levels, and multilocus sequence typing of K. pneumoniae isolates. 33 isolates of K. pneumoniae were isolated and identified in 213 nasal swabs samples, of which 12 were hypervirulent K. pneumoniae strains. Extended Spectrum Beta-Lactamases genes were found in 93.4% of the strains. Of which, TEM was the most prevalent (93.4%), followed by CTX-M and SHV were 57.6% and 39.4%, respectively. A main mutation pattern of quinoloneresistance-determining region, Thr83-Ieu and Asp87-Asn in gyrA and Ser87-Ile in parC, was detected in 33 K. pneumoniae isolates. All the isolates harbored at least two virulence factor genes, with ureA (97.0%) and wabG (91.0%) exhibiting high carriage rates in 33 K. pneumoniae isolates. MLST revealed 7 sequence types, of which 3 STs (2541, 2581 and 2844) were newly assigned. Using eBURST, ST2844 and ST2541 were assigned to new clonal complex 2844. Our study provides evidence and biological characteristics of K. pneumoniae isolates from cattle upper respiratory tract in Southwest China.
Klebsiella pneumoniae (K. pneumoniae) widely existed in surface water, sewage, soil, plants and the mucosal surfaces of mammals.1 Due to its broad spectrum of virulence factors, the infection of K. pneumoniae usually plays an important role for the causing of pneumonia, bloodstream infection and pyogenic liver abscesses in mammals.1,2 The invasion of K. pneumoniae in domestic animals not only causes hazard in livestock production but also poses a potential threat to public health since these animals can act as the reservoir of multidrug-resistant K. pneumoniae strains. Although antibiotic therapy is a widely tool for the treatment of infections caused by K. pneumoniae, however, antibiotic resistance in pathogenic bacteria from food-producing animals and environmental sources is recognized as a global problem for public health.3 Isolates of K. pneumoniae often display drug resistance phenotypes, making difficulty in choosing sensitive antibiotics for treatment.4 The ciprofloxacin is widely used in the treatment of K. pneumoniae infections in recent years, and the increasing resistance to fluoroquinolones among K. pneumoniae has also been reported.5,6 Fluoroquinolones act by inhibiting the action of target enzymes, DNA gyrase and topoisomerase IV, with both enzymes playing a principal role in DNA replication.7 Some researchers think that alterations in the quinoloneresistance-determining region (QRDR) within DNA gyrase (gyrA and gyrB) and topoisomerase IV (parC and parE) are the major mechanisms one for fluoroquinolone resistance in K. pneumoniae.6
Genotyping is important to identify cases or outbreaks due to K. pneumoniae and to further track source and spreading of infections.4 MLST is considered a good method for typing bacterial pathogens and is used to characterize genetic relationships among bacterial isolates and to identify and track the global spread of drug-resistant strains.8–10 It has been shown that the turkey source epidemic clone-related ST15 K. pneumoniae was able to colonize humans and cause severe infections.11 In addition, both handling and consumption of products of these animals colonized by K. pneumoniae may be an important source of antibiotic-resistant K. pneumoniae of possible human health significance.11 However, in contrast to the studies of K. pneumoniae infections in human and other animals,4,12 less is known on the genotyping of K. pneumoniae in cattle or their relevant products. Cattle as an important food-producing animals, it is of importance to understand the prevalence and genotyping characterization of K. pneumoniae colonizing the cattle.
In 2008, Chongqing municipality was incorporated into the national “Advantage of agricultural products regional planning” in China and recognized as the key areas for beef cattle breeding.13 However, the genetic features and microbiological Characteristics of K. pneumoniae isolates from cattle in Southwest China have been poorly investigated. To understand the prevalence and characteristics of K. pneumoniae in cattle, we used the nasal swabs from sick cattle to isolate K. pneumoniae, tested their antimicrobial susceptibility and virulence gene carrying levels, and defined MLST.
Materials and methodsIsolation and identificationDuring the period from 2014 to 2015, 213 nasal swabs samples were collected from 1 to 2 years age of sick cattle suffering from respiratory manifestations. All nasal swab samples were collected from different farms located in Rongchang, Fengdu, Zigong and Yongchuan regions, China. Each sample was plated on LB agar (Oxoid, Thermo Fisher Scientific, China). All plates were incubated at 37°C for 24h. Isolated strains were further investigated by polymerase chain reaction (PCR) using 16S ribosomal RNA primers 27F and 1492R14 with the following amplification conditions: initial denaturation for 5min at 94°C, followed by 30 cycles of 94°C for 1min, 50°C for 1min, and 72°C for 1min, with a final elongation of 72°C for 10min. PCR products were sequenced both in forward and reverse directions by GeneCreate Co., Ltd. (Wuhan, China). The sequencing results of PCR products were analyzed by BLAST (Basic Local Alignment Search Tool) sequence search using the NCBI (National Center for Biotechnology Information) database. Phylogenetic trees were constructed for the 16S rRNA sequences of the isolates by MAFFT Multiple Sequence Alignment and MEGA7.0.17 Characterization of identified K. pneumoniae isolates were further analyzed using MLST, antibiotic resistance and virulence-associated gene profile.
Antimicrobial susceptibility testingThe evaluation of specimens for antibiotic resistance was conducted according to the clinical and laboratory standards institute (CLSI) guidelines.15 Antibiotic susceptibility testing was performed by Kirby–Bauer disk diffusion test. Each sample was tested against 8 antimicrobial agents (Oxoid, Thermo Fisher Scientific, China): Ampicillin (10μg), Amoxicillin (10μg), Gentamicin (10μg), Cefotaxime (30μg), Ceftriaxone (30μg), Ceftazidime (30μg), Ciprofloxacin (10μg) and Amikacin (30μg). The β-lactamase genes (CTX-M, TEM and SHV gene) of the isolates were determined as described by Newire et al.2
Amplification and sequencing of the gyrA and parC fragmentsTo investigate the characteristics of gyrA and parC genes alterations, gyrA and parC gene fragments were amplified and sequenced in 33 isolates. The gyrA and parC primers were used as described by Brisse et al.16 The nucleotide sequences and the deduced amino acid were compared with that of K. pneumoniae strain 459 (GenBank accession number CP018306) using the Clustal W multiple sequence alignment program in MEGA 7.0.17
String test and detection of virulence genesPCR assay were performed to check for the presence of 5 genes that have previously associated with virulence in K. pneumoniae, including magA, rmpA, mrkA, ureA and wabG genes.18,19 Reference strains including ATCC 25922 and ATCC 700603. Strains with the hypermucoviscosity phenotype were defined as high virulent.20 String test was performed to distinguish hypervirulent K. pneumoniae (hvKP) from classic K. pneumoniae (cKP). hvKP phenotype strains was defined by a positive “string test,” which is the formation of a viscous string >5mm in length when bacterial colonies on an agar plate are stretched by an inoculation loop.20
MLST analysisMLST of K. pneumoniae isolates were performed as previously described by Pasteur Institute MLST Database.21 PCR amplification for seven housekeeping genes (gapA, infB, mdh, pgi, phoE, rpoB, and tonB) was carried out using primers and protocols available at the Pasteur Institute and University College Cork scheme described.21 The eBURST v3 software was used to investigate the population diversity and relationship between MLST sequence types (STs) and to analyze clonal complexes (CC) based on the stringent group definition of six out of seven shared alleles. To analyze the phylogenetic relationship of these 33 K. pneumoniae isolates and previously identified K. pneumoniae isolates globally. Along with the STs obtained in this study, 16 STs from human (ST208, 347, 388, 475, 597, 712, 1096 and 1230) and bovine mastitis (ST87, 98, 99, 103, 104, 110, 116 and 1864)4,22 were downloaded from the K. pneumoniae MLST website, concatenated and aligned using MEGA 7.0 and used to construct the NJ phylogenetic tree of K. pneumoniae. Bootstrapping was performed with 1000 replicates.
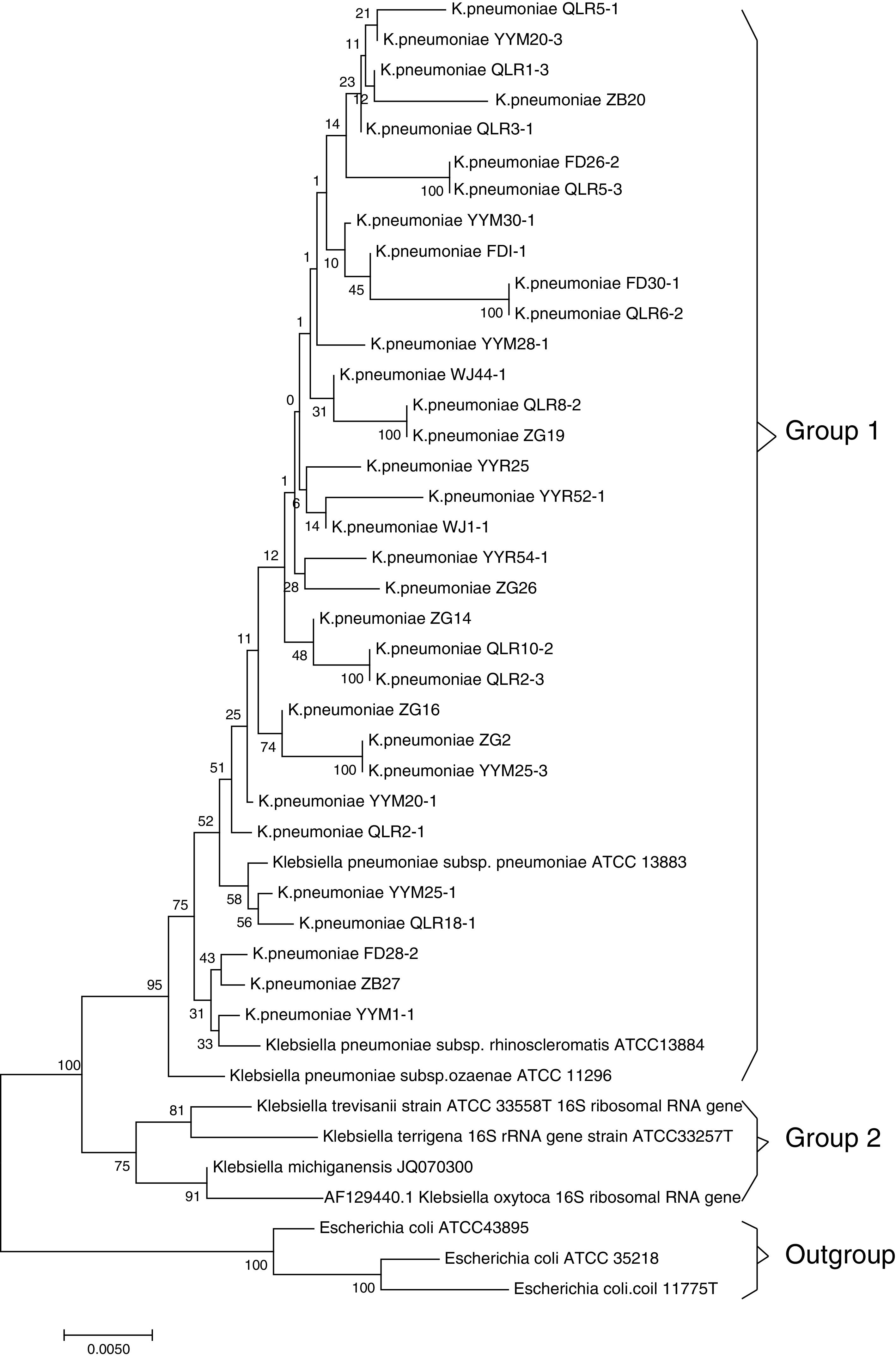
ResultsMicrobiologic characterization of the K. pneumoniae strainsIn this study, 33 (15.5%) strains of K. pneumoniae were detected in the 213 cases investigated by 16S rRNA analysis. The NCBI BLAST program showed that the sequence data of isolated strains had high similarity (>99%) to those of Klebsiella pneumoniae subsp. NTUH-K2044 (AP006725.1). Based on the homology of 16S rRNA sequence, strains with 16S rRNA sequence from relative species were selected, and multiple sequence alignment comparison was performed by MAFFT-LINSI program. A phylogenetic tree was constructed using the NJ method based on MEGA 7.0 software. The results of phylogenetic placement of 33 isolates were shown in Fig. 1. These isolates were mainly clustered in Klebsiella pneumoniae subsp (Group 1). The gene sequence of 16S rRNA was deposited in the NCBI nucleotide sequence database under accession No. MF767570-MF767596, MG859650-MG859655.

Phylogenetic tree of the K. pneumoniae based on 16S rRNA sequences. Note: The 33 sequences determined in this study are indicated by bold font. 3 Escherichia coli strains, Z83205.1, AM980865.1 and X80725.1, were used as outgroups; Bootstrap values of 1000 replications are indicated at branches. The sequences in group 1 correspond to the clusters in Klebsiella pneumoniae subsp.
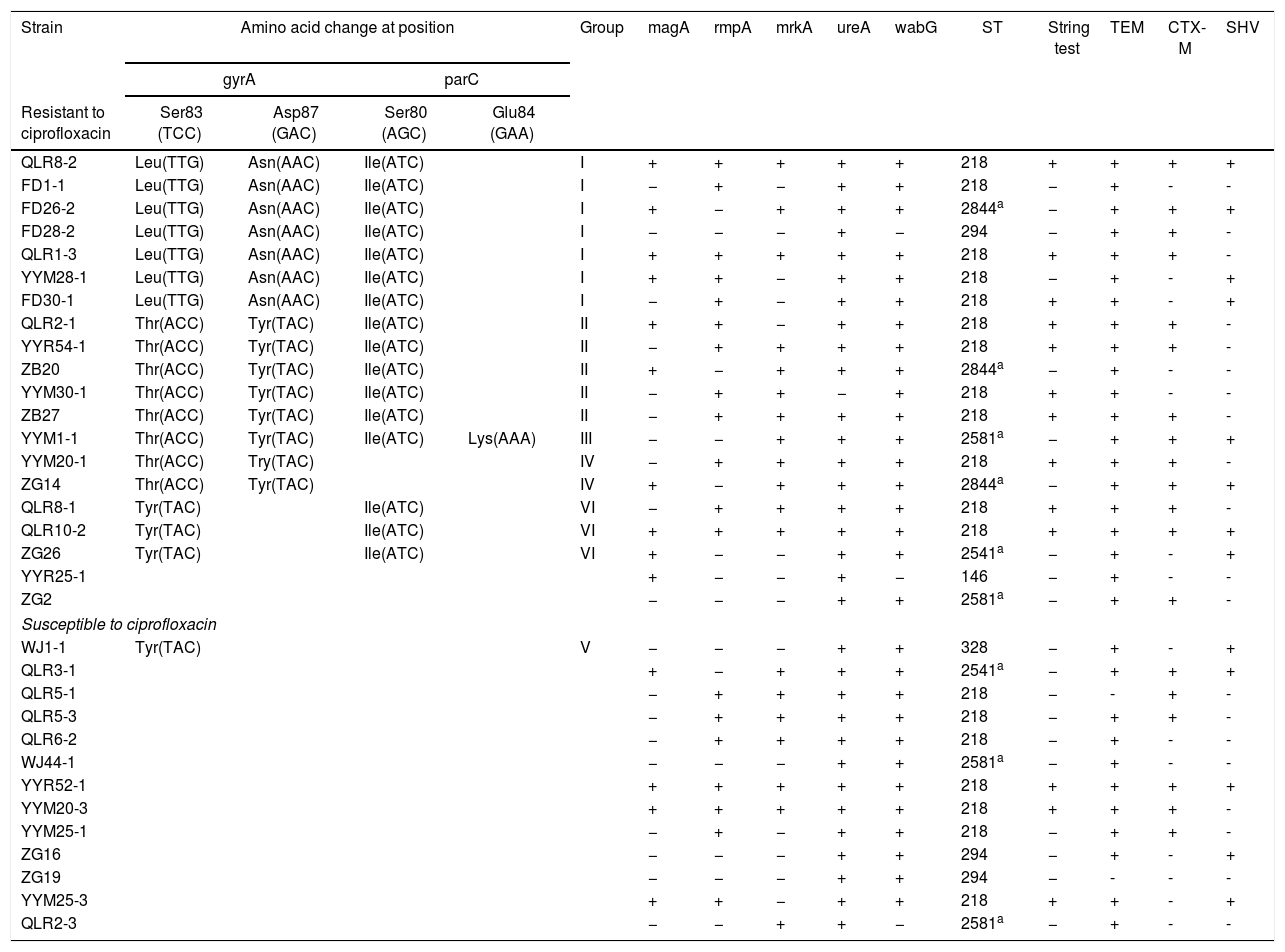
33 K. pneumoniae isolates resistance rates to Ampicillin, Amoxicillin, Ciprofloxacin, Cefotaxime, Ceftriaxone, Gentamicin, Ceftazidime and Amikacin were 93.9%, 81.8%, 60.6%, 57.6%, 33.3%, 27.3%, 18.2% and 9.1%, respectively. Extended Spectrum Beta-Lactamases (ESBLs) genes were found in 93.4% of the strains. Of which, TEM was the most common (93.4%), followed by CTX-M and SHV were 57.6% and 39.4%, respectively (Table 1). Among 33 isolates, 19 were revealed to have amino acid alterations in gyrA or parC QRDR (Table 1). The total mutations found in these isolates were classified into 6 distinct groups according to the pattern of amino acid alteration, in which group I showed the most common in 33 isolates. It is notable that all isolates in group 1 showed ciprofloxacin resistance characteristics, which may indicate that this mutation pattern is a suitable model for K. pneumoniae resistance to ciprofloxacin. Of six groups, only group III had two mutations in gyrA (Ser83-Thr; Asp87-Tyr) and parC (Ser80-Ile; Glu84-Lys), respectively. DNA sequences of QRDR gyrA showed a double mutation in gyrA was detected in 15 of 33 isolates. The sequences of the gyrA and parC genes in clinical isolates of K. pneumoniae have been submitted to the GenBank database under accession numbers MG758055-MG758108; MG869692-MG869703.
Molecular characteristics of Klebsiella pneumoniae isolates.
| Strain | Amino acid change at position | Group | magA | rmpA | mrkA | ureA | wabG | ST | String test | TEM | CTX-M | SHV | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gyrA | parC | ||||||||||||||
| Resistant to ciprofloxacin | Ser83 (TCC) | Asp87 (GAC) | Ser80 (AGC) | Glu84 (GAA) | |||||||||||
| QLR8-2 | Leu(TTG) | Asn(AAC) | Ile(ATC) | I | + | + | + | + | + | 218 | + | + | + | + | |
| FD1-1 | Leu(TTG) | Asn(AAC) | Ile(ATC) | I | − | + | − | + | + | 218 | − | + | - | - | |
| FD26-2 | Leu(TTG) | Asn(AAC) | Ile(ATC) | I | + | − | + | + | + | 2844a | − | + | + | + | |
| FD28-2 | Leu(TTG) | Asn(AAC) | Ile(ATC) | I | − | − | − | + | − | 294 | − | + | + | - | |
| QLR1-3 | Leu(TTG) | Asn(AAC) | Ile(ATC) | I | + | + | + | + | + | 218 | + | + | + | - | |
| YYM28-1 | Leu(TTG) | Asn(AAC) | Ile(ATC) | I | + | + | − | + | + | 218 | − | + | - | + | |
| FD30-1 | Leu(TTG) | Asn(AAC) | Ile(ATC) | I | − | + | − | + | + | 218 | + | + | - | + | |
| QLR2-1 | Thr(ACC) | Tyr(TAC) | Ile(ATC) | II | + | + | − | + | + | 218 | + | + | + | - | |
| YYR54-1 | Thr(ACC) | Tyr(TAC) | Ile(ATC) | II | − | + | + | + | + | 218 | + | + | + | - | |
| ZB20 | Thr(ACC) | Tyr(TAC) | Ile(ATC) | II | + | − | + | + | + | 2844a | − | + | - | - | |
| YYM30-1 | Thr(ACC) | Tyr(TAC) | Ile(ATC) | II | − | + | + | − | + | 218 | + | + | - | - | |
| ZB27 | Thr(ACC) | Tyr(TAC) | Ile(ATC) | II | − | + | + | + | + | 218 | + | + | + | - | |
| YYM1-1 | Thr(ACC) | Tyr(TAC) | Ile(ATC) | Lys(AAA) | III | − | − | + | + | + | 2581a | − | + | + | + |
| YYM20-1 | Thr(ACC) | Try(TAC) | IV | − | + | + | + | + | 218 | + | + | + | - | ||
| ZG14 | Thr(ACC) | Tyr(TAC) | IV | + | − | + | + | + | 2844a | − | + | + | + | ||
| QLR8-1 | Tyr(TAC) | Ile(ATC) | VI | − | + | + | + | + | 218 | + | + | + | - | ||
| QLR10-2 | Tyr(TAC) | Ile(ATC) | VI | + | + | + | + | + | 218 | + | + | + | + | ||
| ZG26 | Tyr(TAC) | Ile(ATC) | VI | + | − | − | + | + | 2541a | − | + | - | + | ||
| YYR25-1 | + | − | − | + | − | 146 | − | + | - | - | |||||
| ZG2 | − | − | − | + | + | 2581a | − | + | + | - | |||||
| Susceptible to ciprofloxacin | |||||||||||||||
| WJ1-1 | Tyr(TAC) | V | − | − | − | + | + | 328 | − | + | - | + | |||
| QLR3-1 | + | − | + | + | + | 2541a | − | + | + | + | |||||
| QLR5-1 | − | + | + | + | + | 218 | − | - | + | - | |||||
| QLR5-3 | − | + | + | + | + | 218 | − | + | + | - | |||||
| QLR6-2 | − | + | + | + | + | 218 | − | + | - | - | |||||
| WJ44-1 | − | − | − | + | + | 2581a | − | + | - | - | |||||
| YYR52-1 | + | + | + | + | + | 218 | + | + | + | + | |||||
| YYM20-3 | + | + | + | + | + | 218 | + | + | + | - | |||||
| YYM25-1 | − | + | − | + | + | 218 | − | + | + | - | |||||
| ZG16 | − | − | − | + | + | 294 | − | + | - | + | |||||
| ZG19 | − | − | − | + | + | 294 | − | - | - | - | |||||
| YYM25-3 | + | + | − | + | + | 218 | + | + | - | + | |||||
| QLR2-3 | − | − | + | + | − | 2581a | − | + | - | - | |||||
Note: +: positive reaction; −: negative reaction.
The ureA genes were detected in majority (97.0%) of the strains, and wabG gene was found in 30 (91.0%), followed by mrkA and rmpA in 57.6% isolates each, magA (42.4%). According to the string test results, 12 hvKP strains were identified in the 33 isolates (Table 1).
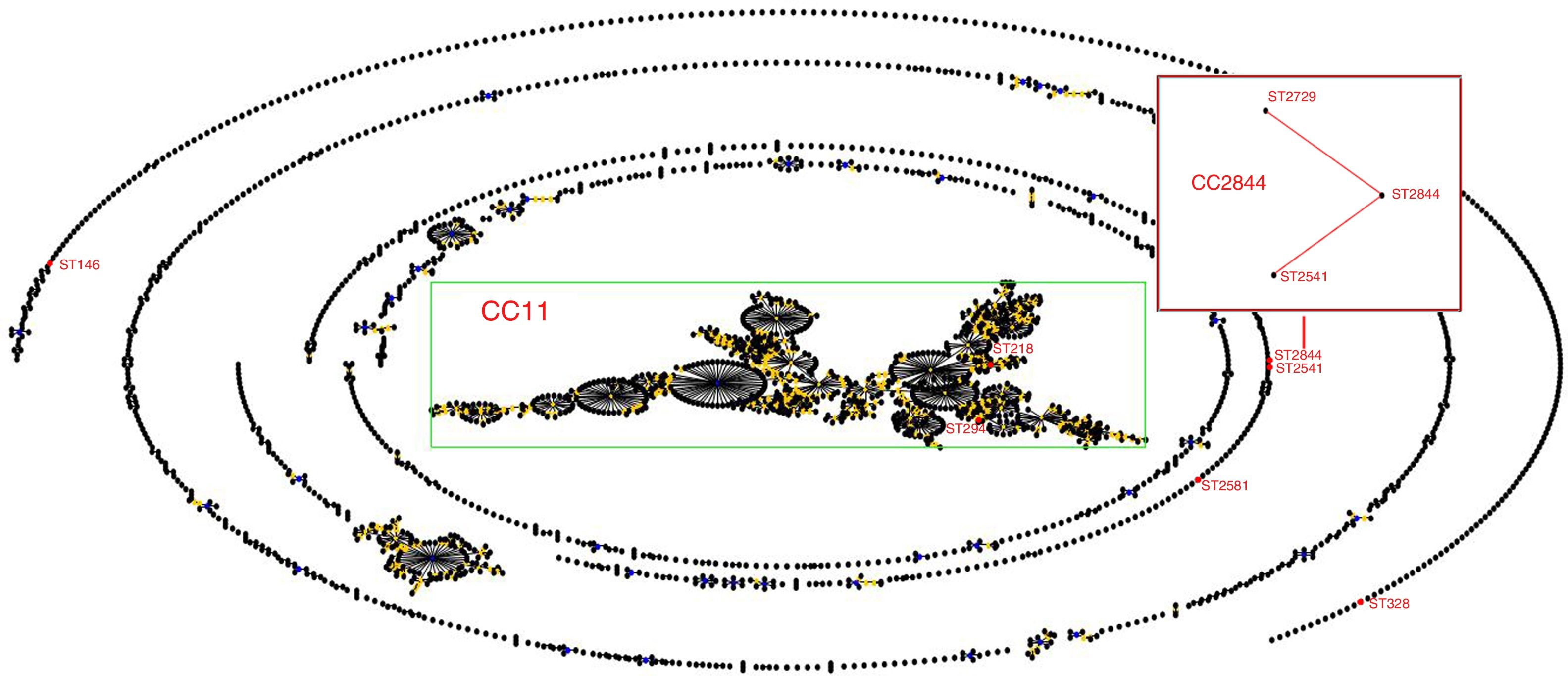
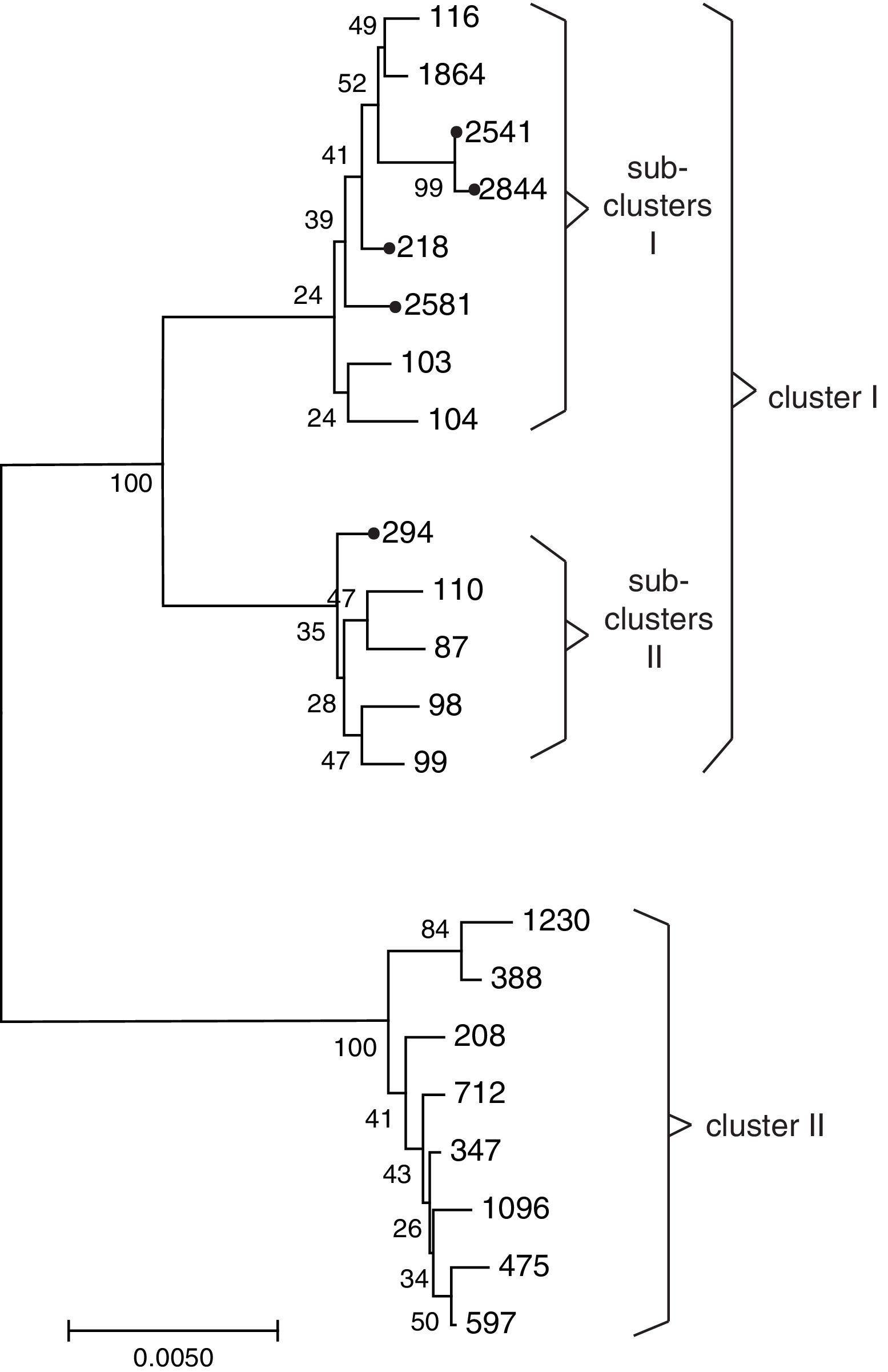
Sequence types of K. pneumonia strainsIn this experiment, a total of 7 STs were found in the 33 K. pneumoniae strains, of which 3 STs were newly assigned. The 4 known MLST genotypes were identified: ST146 (n=1), ST294 (n=3), ST328 (n=1), ST218 (n=19). The 3 newly MLST genotypes were identified and added to the K. pneumoniae MLST database: ST2844 (3-2-4-181-9-4-177; n=3), ST2541 (3-2-4-1-9-4-177; n=2), ST2581 (10-1-1-14-26-1-12; n=4). eBURST generated 232 groups and 923 singletons (Until January 9, 2018). The K. pneumoniae isolates tested in this study were clustered into CC11 containing ST294 and ST218, and a further CC2844 containing ST2844 and ST2541, while ST146, ST328 and ST2581 was a singleton (Fig. 2). The phylogenetic relationship of 21 STs, based on concatenated sequences of seven MLST alleles, resulted in two main clusters: cluster I (mainly from bovine) and cluster II (from humans). In cluster I, there were two sub-clusters (SCs) (Fig. 3). Four K. pneumoniae isolates from CURT were clustered to SC1, and SC 2 mainly contains of the clinical mastitis isolates from cow.
 in this study are indicated by red font.")
 based on neighbor-joining method. Note: The 5 STs determined in this study are indicated by black dot.")
Many reports suggest that K. pneumoniae is one of the major pathogens of bovine mastitis.23–25 However, there is very little information about K. pneumoniae isolates from the upper respiratory tract of cattle. In the study, 33 (15.5%) K. pneumoniae strains were isolated from the upper respiratory tract of cattle. Our results were higher to the detection frequency of K. pneumoniae in Zimbabwe,23 but lower than that in Turkey.26
The majority of 33 K. pneumoniae isolates showed relatively low resistance to Ceftazidime and Amikacin, which is similar to the resistance of K. pneumoniae isolated from patients with hospital-acquired pneumonia in Asian.27 In this study, it is noticeable that K. pneumoniae isolates showed relatively high resistance to ciprofloxacin, which is higher than the resistance of K. pneumoniae from patient in Iran and China.28,29 The prolonged use of antibiotics in the treatment of respiratory illness and as growth promoters will led to the additional problem of emergence antibiotic resistant strains, for example the resistant strains entering the food chain. Therefore, the high resistance strain of ciprofloxacin may be a potential public health hazard in china. Several reports have shown5,6 that changes in the structure of the QRDRs in gyrA and parC are one of the significant mechanisms in conferring a resistance to fluoroquinolone in gram negative bacilli. The mutations of Ser83 with Phe, Ile, Tyr and Leu is frequently displayed in K. pneumoniae in some studies.5,30 However, the results from this study suggested the mainly existence of Ser83-Thr alteration in K. pneumoniae isolates, which may be simply due to the investigation of a new population, since most reports have focused on human clinical isolates or bovine mastitis. In this study, it was found that in the Ciprofloxacin-resistant K. pneumoniae, the mutation of the 80th site in the bacterial parC gene was commonly accompanied by the mutation of the gyrA gene, and when parC gene and gyrA gene mutated the sites together, the Ciprofloxacin-resistance rate increased significantly, the results indicating that mutations of parC gene and gyrA gene will make K. pneumoniae more highly resistant to Ciprofloxacin. DNA sequencing of gyrA and parC in clinical strains has revealed some mutations in the QRDR associated with fluoroquinolone resistance. However, QRDRs of the two isolates (YYR25-1 and ZG2) with resistance to ciprofloxacin in this study did not possess alterations associated with fluoroquinolones resistance in the sequence of either genes, indicating that other unknown resistance mechanisms contribute to K. pneumoniae became resistant to Ciprofloxacin, such as efflux systems and biofilms should be considered in these isolates.
In this study, most of the isolates harbored at least two virulence factor genes, suggesting that CURT K. pneumoniae isolates in this region were potentially pathogenic. Majority of studied isolates were positive for ureA (97.0%) and wabG (91.0%) gene, similarly high prevalence of these genes observed in isolate of human clinical and other animals,19,20 this result indicates that the ureA and wabG genes are prevalent in K. pneumoniae. The type 3 fimbrial shaft mrkA gene detected in 57.6% of isolates, which is less than that 100% reported earlier by Alcantar-Curiel et al.18 This result shows that there are differences between bovine isolates and human clinical isolates. Many reports suggest that the rmpA-carrying strains were associated with the hypermucoviscosity phenotype. In this study, the detection rates of rmpA and magA were 57.6% and 42.4%, respectively, and string test results revealed that 36.4% K. pneumoniae isolates from cattle showed hypermucoviscosity. The hvKP rate was significantly lower than that human clinical isolates (85%).29 However, the string test result is lower than the rmpA gene detection rate, which indicate that string test might not be a reliable method to identify hvKP.
In this study, ST218 was the most common, which occurred in approximately 57.6% of the K. pneumonia isolates. Through MLST database and some research reports27,31 that ST218 was isolated from human in Thailand, Vietnam, South Korea and Russia, and Liao et al.31 reported that ST218 strains may cause liver abscess disease in human. ST218 is closely related to the ST23 prevailing in Europe and the United States, both of them belongs to CC11, and only a housekeeping gene (tonB) difference between each other. The ST23 has caused serious public health events in many parts of the world.1 As mentioned above, the prevalence of ST218 isolates in cattle is not negligible. Three new STs (ST2541, 2581 and 2844) were sub-grouped in the branch of SC1 containing ST218 and ST116. Previous research revealed that ST116 can cause bovine mastitis.22 Our works confirmed the relative close relationship of ST2541, ST2581 and ST2844 with ST218 and ST116. On the other hand, the distribution of virulence factors among the different sequence types was not random. In this study, isolates belonging to ST218 were with the genes mrkA+ureA+wabG, whereas ST2844 included isolates with rmpA+wabG genes. This is the first study to report the frequency, antimicrobial resistance profile, virulence genes and MLST molecular characteristic of K. pneumoniae from cattle in Southwest China. This study will help continuous monitoring of the genetic diversity of K. pneumoniae may disclose differences in virulence, epidemiology, evolution and genetic traits that influence control and treatment.
Conflicts of interestThe authors declared no potential conflicts of interest with respect to the research, authorship, and publication of this article.
This study was supported by the Fundamental Research Funds for the Central Universities (XDJK2017D083), Innovation Project for the Social Undertakings and People's Livelihood Protection in Chongqing (CSTC2015SHMSZX80020) and Frontiers and basic research projects in Chongqing (cstc2016jcyA0235).