







Small ruminant lentiviruses (SRLV) have high genetic variability which results in different viral strains around the world. This create a challenge to design sensible primers for molecular diagnosis in different regions. This work proposes a protocol of duplex nested-PCR for the precise diagnosis of SRLV. The technique was designed and tested with the control strains CAEV Co and MVV 1514. Then, field strains were submitted to the same protocol of duplex nested-PCR. Blood samples of sheep and goats were tested with AGID and nested PCR with specific primers for pol, gag and LTR. The AGID results showed low detection capacity of positive animals, while the nested PCR demonstrated a greater capacity of virus detection. Results demonstrated that LTR-PCR was more efficient in detecting positive sheep samples, whereas gag-PCR allowed a good detection of samples of positive goats and positive sheep. In addition, pol-PCR was more efficient with goat samples than for sheep. Duplex nested PCR performed with standard virus samples and field strains demonstrated that the technique is more efficient for the detection of multiple pro-viral DNA sequences. This study demonstrated a successful duplex nested PCR assay allowing a more accurate diagnosis of SRLV.
Small ruminant lentiviruses (SRLV) are pathogens that affect sheep and goats causing persistent and progressive infections. These agents are represented by two phylogenetic groups characterized by the Maedi-Visna virus (MVV) and the caprine arthritis encephalitis virus (CAEV).1–3 Phylogenetic analysis divides these viruses into five major groups classified as A-E. The MVV and CAEV prototypes of groups A and B, respectively, are widely distributed throughout the world, while C-E groups are geographically restricted.4,5
These lentiviruses are closely related and were considered species-specific infecting sheep and goats, respectively, causing progressive lung disease, encephalomyelitis, mastitis and arthritis.6 However, molecular and epidemiological studies conducted in the last decades indicate that these agents are capable of infecting both goats and sheep,7 transgressing the barrier between species consistently and easily.4,8,9
These viruses present high genetic variability related to the viral replication process, resulting in the formation of several viral strains in different geographic regions.2 Due to the heterogeneity of the SRLV genome it is difficult to use the same set of primers in different geographic regions.10
The serological diagnosis of infected animals by the Agarose Gel Immunodiffusion (AGID) technique is widely used and accepted due to its low cost and practicality. 11 In addition, this technique is recommended by OIE.12 However, some animals may present low antibody titers, late seroconversion or intermittent reactions of seropositivity and seronegativity.13,14
Numerous studies have successfully demonstrated the use of the PCR technique in the detection of pro-viral DNA from SRLV15–18 due to the high sensitivity and specificity this technique presents.19 A multiplex PCR is a variant of the conventional PCR in which two or more target sequences can be amplified including more than one pair of primers in the same reaction.20
Based on these advances of molecular diagnosis, this study aimed to develop a duplex nested PCR protocol for accurate diagnosis of small ruminant lentiviruses, as well as to verify the efficiency of the method in diagnosing sheep and goat field samples.
Material and methodsThis project was approved by the local Ethics Committee for the Use of Animals from the State University of Ceará, protocol number 0784899-15.
SamplesFor testing duplex nested-PCR technique, standard samples of the CAEV-Cork21 and MVV-K151422 strains were used and this virus samples were amplified in cell culture and stored frozen for later use.
These samples were provided by Dr. Roberto Soares de Castro (Federal Rural University of Pernambuco – UFRPE) and originated from the Institut National de la Recherche Agronomique Lyon-France.
To demonstrate the efficiency of the technique in detecting SRLV, samples were collected from animals housed in two farms of northeastern mesoregion from Pará state and two farms in the metropolitan region of Fortaleza, Ceará – Brazil. Herds of Santa Inês breed and undefined breed were used in this study. Similarly, samples of goats were obtained from different properties in metropolitan region of Fortaleza, Ceará – Brazil. Herds of Saanen, Alpina, Murciana breed and crossbred animals were collected. All animals were aged from 1 to 2 years and the findings of clinical symptomatology were described in Table 2.
Blood samples of 20 goats and 15 sheep were collected by venipuncture of the jugular vein using vacutainer® tubes with EDTA to perform molecular tests and tubes without anticoagulant for agarose gel immunodiffusion (AGID) assay.
Agarose gel immunodiffusion assay (AGID)Before performing duplex nPCR in field strains, all samples were tested by AGID for screening animals and identifying positives and negatives. AGID test was performed using the national commercial kit developed by Biovetech®. The above-mentioned AGID kit is produced using supernatant concentrate antigen from MVV and CAEV infected cell cultures and with positive serum harvested from naturally infected animal. This test is indicated for detection of anti-p28 antibodies from goats infected with CAEV and sheep infected with MVV.
DNA extractionFor DNA extraction from whole blood leukocytes, samples were submitted to the protocol described by Caldas et al.23. The samples were centrifuged at 1500g for 10min. The leukocyte layer was collected and transferred to 1.5μL microtubes containing 500μL TE (10mM Tris, 1mM). The material was centrifuged in microfuge at 2700g for 5min to obtain the pellet, then the supernatant was discarded and the operation repeated. The pellet was resuspended in K buffer (10mM Tris HCl pH 8.0, 50mM KCl, 2mM MgCl2, 0.5% Tween 20, 100μg/mL proteinase K=0.1μg/μL). The samples were vortexed, incubated at 56° C for 45min and then boiled at 96° C for 10min. The material was stored at 4° C for 24h for a better homogenization of sample and then at −20°C until duplex nPCR was performed.
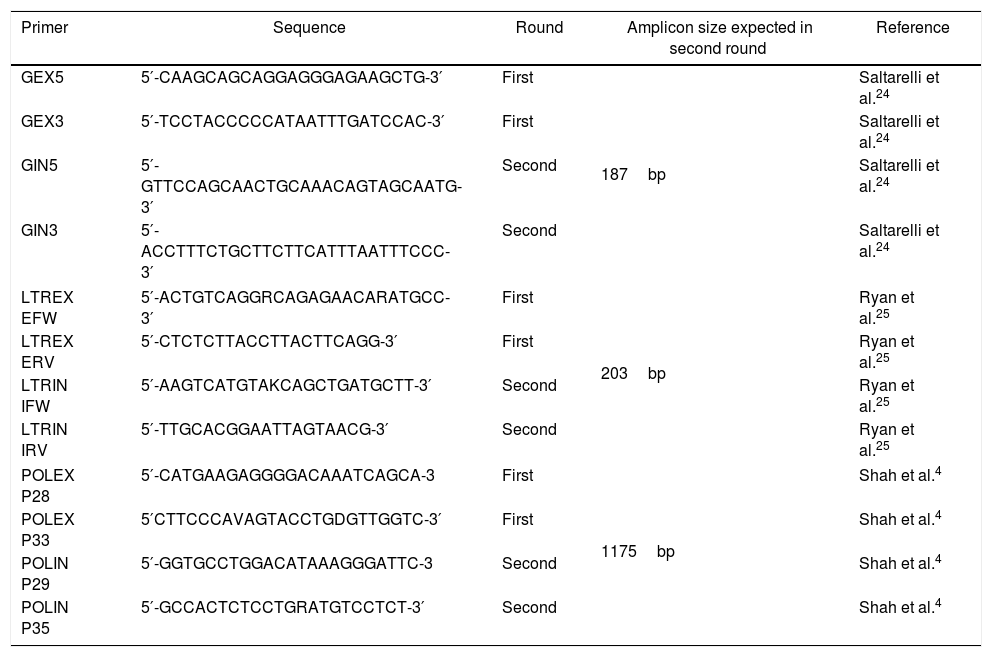
PrimersFor duplex nPCR, a pair of primers was used for each region (gag, pol, LTR), which are located in more conserved regions (Table 1).
Primers used for duplex nested-PCR standardization.
| Primer | Sequence | Round | Amplicon size expected in second round | Reference |
|---|---|---|---|---|
| GEX5 | 5′-CAAGCAGCAGGAGGGAGAAGCTG-3′ | First | 187bp | Saltarelli et al.24 |
| GEX3 | 5′-TCCTACCCCCATAATTTGATCCAC-3′ | First | Saltarelli et al.24 | |
| GIN5 | 5′-GTTCCAGCAACTGCAAACAGTAGCAATG-3′ | Second | Saltarelli et al.24 | |
| GIN3 | 5′-ACCTTTCTGCTTCTTCATTTAATTTCCC-3′ | Second | Saltarelli et al.24 | |
| LTREX EFW | 5′-ACTGTCAGGRCAGAGAACARATGCC-3′ | First | 203bp | Ryan et al.25 |
| LTREX ERV | 5′-CTCTCTTACCTTACTTCAGG-3′ | First | Ryan et al.25 | |
| LTRIN IFW | 5′-AAGTCATGTAKCAGCTGATGCTT-3′ | Second | Ryan et al.25 | |
| LTRIN IRV | 5′-TTGCACGGAATTAGTAACG-3′ | Second | Ryan et al.25 | |
| POLEX P28 | 5′-CATGAAGAGGGGACAAATCAGCA-3 | First | 1175bp | Shah et al.4 |
| POLEX P33 | 5′CTTCCCAVAGTACCTGDGTTGGTC-3′ | First | Shah et al.4 | |
| POLIN P29 | 5′-GGTGCCTGGACATAAAGGGATTC-3 | Second | Shah et al.4 | |
| POLIN P35 | 5′-GCCACTCTCCTGRATGTCCTCT-3′ | Second | Shah et al.4 | |
Before performing duplex nPCR in field samples, all samples were tested by nested-PCR (nPCR) for screening the animals and identifying positives and negatives. The detection of pro-viral DNA was performed with nPCR following the methodology described by Barlough et al.26 with modifications. Modifications were made to the methodology as proposed by Andrioli et al.27 in which KCl, gelatin and tetramethylammonium chloride were removed from the PCR mix. In addition, modifications were made to the amplification cycles with a decrease in the number from 45 to 35 cycles and an increase in the annealing temperature from 50°C to 63°C.
This was performed to amplify a fragment of the CAEV and MVV pro-viral DNA based on the gag, pol and LTR regions adjusting the of primer concentration. Individual nPCR was performed for each set of primers, and all samples were tested for the gag, pol and LTR primers prior to performing the duplex nPCR. The nPCR was performed with gag primers were considered as standard to discriminate positive or negative samples for the molecular tests. This choice was made on the basis of findings from the literature confirming that the gag region is the most conserved within the SRLV genome.37,41–43
The reactions was performed in a thermal cycler (Eppendorf®) in a final volume of 50μL and the solution mix consisted of 10mM Tris HCl buffer (pH 8.3); 1.5mM MgCl2; 100μM of each dNTP; 2U/μL taq DNA polymerase recombinant (Invitrogen®); 3μL of test sample and DNAse-free water to complete a final volume. Different concentrations of the gag, pol and LTR primers prepared for each reaction were tested with the concentrations of 20pM, 10pM, 7.5pM and 5pM.
The amplification conditions were as follows: initial denaturing at 94°C for five min, followed by 35 cycles: denaturation at 94°C for one min, annealing 63°C for one min, extension at 72°C for 45s, followed by an additional extension phase at 72°C for seven min. An aliquot of 1.0μL of this PCR product was transferred directly to a second reaction. The first and second rounds of the PCR were performed under the same previous conditions. All reactions were performed under the same time and temperature conditions.
PCR amplified fragments were analyzed by horizontal cube electrophoresis and the bands were visualized under the ultraviolet light transilluminator (Vilber Lourmat – Transilluminator UV/white light) and photodocumented (Logic 200 PRO Serie). The sizes of the amplified fragments were compared to a 100bp DNA ladder (Promega) and to the positive control.
Duplex nested-PCR using standard SRLV samplesAfter the serological test and the individual nPCR performed with each set of primers, the duplex nPCR was performed with the same primer concentrations of the previous test. The following concentrations were used 20pM, 10pM, 7.5pM and 5pM for gag, pol and LTR primers.
The first step of standardization involved performing the duplex nPCR using the primers for the gag and pol genes and the second step involved a test using the primers for the LTR and pol region.
The combination gag and LTR was not evaluated. Primers designed for this combination did not generate satisfactory results in the tests performed, since the amplicons generated have very close sizes, making it impossible to differentiate them.
The reactions were performed in a thermal cycler (Eppendorf®) in a final volume of 50μL and the solution mix consisted of 10mM Tris HCl buffer (pH 8.3); 2U/μL taq DNA polymerase; 3μL of test sample and DNAse-free water to complete a final volume. The primer concentrations were used according to the results obtained for the conventional nPCR previously performed. To adjust the duplex nPCR technique, different concentrations of dNTPs and magnesium were also tested. Amplification conditions followed the same protocol as previously described for the nPCR.
PCR amplified fragments were analyzed by horizontal cube electrophoresis. Different concentrations of agarose gel were tested to visualize PCR products, in which the following were tested 1%, 1.3%, 1.5% and 2%. The concentration that allowed the best visualization of the different bands formed in the duplex nPCR was verified. The bands were visualized in ultraviolet light transilluminator (Vilber Lourmat – Transilluminator UV/white light) and photodocumented (Logic 200 PRO Serie). The sizes of the amplified fragments were compared to 100bp DNA ladder (Promega) and to the positive control.
Evaluation of technique performance in field samplesAfter obtaining the best concentration of reagents for duplex nPCR and evaluating effectiveness in detecting the standard virus samples, the developed technique was used to test the samples obtained from animals in the field.
Fifteen sheep and twenty goat samples were tested with duplex nPCR using the pol and gag primer sets and the pol and LTR primer sets as described for the experiment performed with the standard virus samples.
DNA sequencing and sequence analysisPositive samples obtained from Goats 6, 7 and 8 and Sheep 4, 5 and 6 were sent for sequencing.
The partial direct sequencing of the gag gene relative to the amplification products obtained in the second round of nested-PCR was performed. Were used the primers internals GIN5 and GIN3.
The samples were purified on 1% agarose gel using commercial kit (BigDye Terminator v3.1 Cycle Sequencing) and sequenced on ABI 3500 platform (Genetic Analyzer). The gene sequences were compared with MEGA7 analysis software, and Basic Local Alignment Search Tool – Blast. All positions with ambiguous codes or alignment gaps were excluded from the analyses.
The SRLV strains were compared to K1514-MVV,28 CAEV,24 7-29Ont,29 Br/UFRGS-2/V27,30 21G-MG/Br,31 39QPI/Br,31 19F-MG/Br31 and BR/CNPC-G432 with GenBank accession no. M60609, M33677, KC155690.1, AJ305039.1, KF861573.1, KF861553.1, EU300979.1 and KF861559.1 respectively.
Through the analysis by BLASTN 2.8.0 tool it was possible to determine the similarity of the strains found with the other strains deposited in the GenBank.33,34
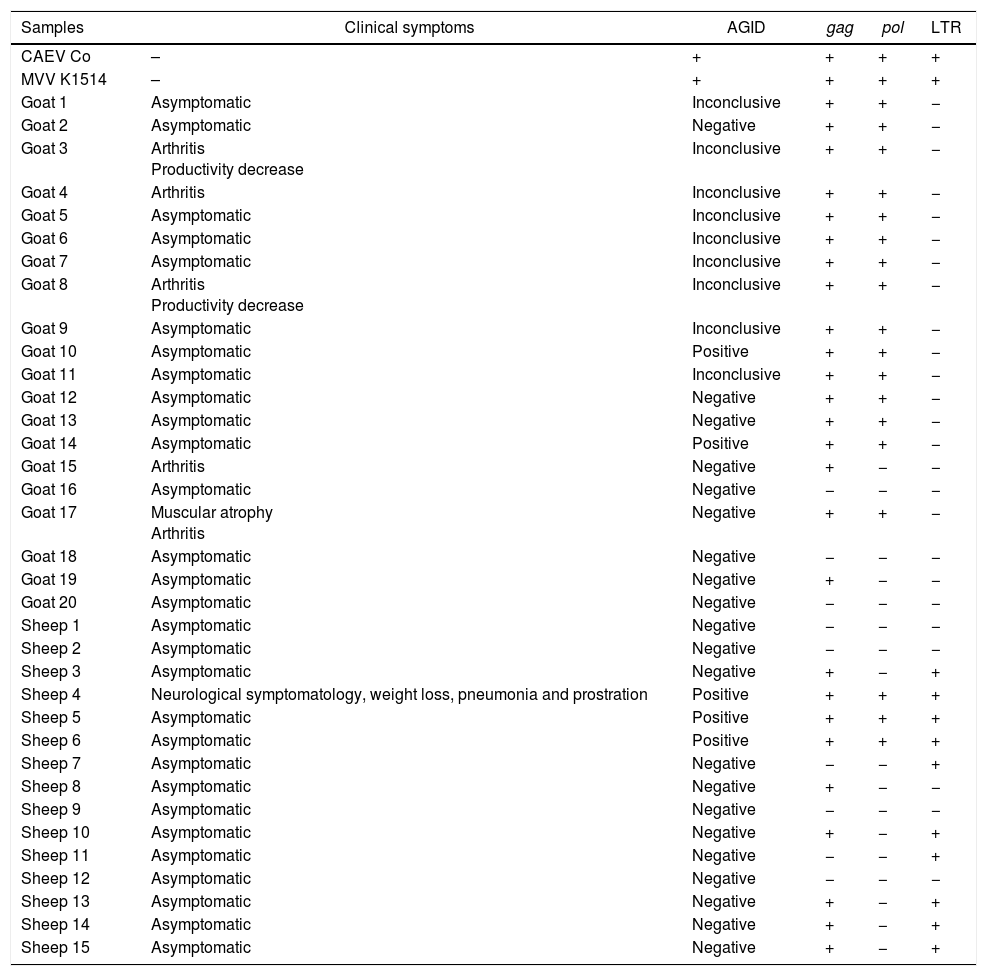
ResultsAGID and nested PCRAll field samples were tested with AGID and nested-PCR techniques, in which the primers for the gag gene were used and considered the diagnostic control standards for comparison with the developed duplex nested-PCR technique. In a total of 20 goats evaluated by AGID, only two animals (10%) were positive, while nine animals had inconclusive. Inconclusive results consisting in inadequate formation of the expected precipitation line.
Same samples were tested with nPCR with gag primers, in which 17 goats (85%) tested positive. Only three animals that were negative in AGID test continued to demonstrate negative results with nPCR performed with gag primers.
In a total of 15 sheep evaluated by AGID, three presented positive results and 12 were negative. In the nPCR results using gag primers, 9 (60%) sheep were positive and 6 sheep with negative results in AGID were also negative in nPCR.
As for the technique performed with the pol primers, results demonstrated that primers were also efficient for detecting pro-viral DNA from goat samples, but not as efficient as primers for gag region. Among the 17 animals positive in nPCR performed with gag, 15 animals (75%) were also positive for the pol primers, showing an agreement of 88%. However, the nPCR results of sheep samples using pol primers demonstrated that these were not as efficient in detecting the positive animals. From the total of fifteen tested animals, only 3 were positive for the technique using the pol primers (Table 2).
Description of the clinical findings and the results obtained in the AGID and Nested-PCR tests.
| Samples | Clinical symptoms | AGID | gag | pol | LTR |
|---|---|---|---|---|---|
| CAEV Co | – | + | + | + | + |
| MVV K1514 | – | + | + | + | + |
| Goat 1 | Asymptomatic | Inconclusive | + | + | − |
| Goat 2 | Asymptomatic | Negative | + | + | − |
| Goat 3 | Arthritis Productivity decrease | Inconclusive | + | + | − |
| Goat 4 | Arthritis | Inconclusive | + | + | − |
| Goat 5 | Asymptomatic | Inconclusive | + | + | − |
| Goat 6 | Asymptomatic | Inconclusive | + | + | − |
| Goat 7 | Asymptomatic | Inconclusive | + | + | − |
| Goat 8 | Arthritis Productivity decrease | Inconclusive | + | + | − |
| Goat 9 | Asymptomatic | Inconclusive | + | + | − |
| Goat 10 | Asymptomatic | Positive | + | + | − |
| Goat 11 | Asymptomatic | Inconclusive | + | + | − |
| Goat 12 | Asymptomatic | Negative | + | + | − |
| Goat 13 | Asymptomatic | Negative | + | + | − |
| Goat 14 | Asymptomatic | Positive | + | + | − |
| Goat 15 | Arthritis | Negative | + | − | − |
| Goat 16 | Asymptomatic | Negative | − | − | − |
| Goat 17 | Muscular atrophy Arthritis | Negative | + | + | − |
| Goat 18 | Asymptomatic | Negative | − | − | − |
| Goat 19 | Asymptomatic | Negative | + | − | − |
| Goat 20 | Asymptomatic | Negative | − | − | − |
| Sheep 1 | Asymptomatic | Negative | − | − | − |
| Sheep 2 | Asymptomatic | Negative | − | − | − |
| Sheep 3 | Asymptomatic | Negative | + | − | + |
| Sheep 4 | Neurological symptomatology, weight loss, pneumonia and prostration | Positive | + | + | + |
| Sheep 5 | Asymptomatic | Positive | + | + | + |
| Sheep 6 | Asymptomatic | Positive | + | + | + |
| Sheep 7 | Asymptomatic | Negative | − | − | + |
| Sheep 8 | Asymptomatic | Negative | + | − | − |
| Sheep 9 | Asymptomatic | Negative | − | − | − |
| Sheep 10 | Asymptomatic | Negative | + | − | + |
| Sheep 11 | Asymptomatic | Negative | − | − | + |
| Sheep 12 | Asymptomatic | Negative | − | − | − |
| Sheep 13 | Asymptomatic | Negative | + | − | + |
| Sheep 14 | Asymptomatic | Negative | + | − | + |
| Sheep 15 | Asymptomatic | Negative | + | − | + |
The nPCR performed with the LTR primers demonstrated that these primers were not efficient for the detection of pro-viral DNA in goat samples of this study, since none of the tested animals presented a positive result. Among the 15 sheep samples tested for this set of primers, 10 were positive. One sample that had been considered negative for the pol and gag primers showed positivity for the LTR primers.
The best primer concentrations for nPCR in this study were 10pM for gag primers and 20pM for LTR. For pol primers the best concentration was 7.5pM per reaction. For this test, there was no need to modify the concentration of any other reagent or adjust the thermal cycler.
Duplex nested-PCR standardization using standard SRLV samplesFor standardization of the duplex nPCR technique, tests were elaborated with standard samples of CAEV-Cork and MVV-K1514 viruses. This step was performed in order to demonstrate that the primers prepared and used for performing the technique are effective in detecting the SRLV.
Standardization of the duplex nPCR technique demonstrated that the optimal concentrations for the reaction using the gag and pol primers was 5pM for pol and 10pM for the gag, which generated two satisfactory bands both allowing a conclusive and reliable diagnosis.
For the protocol of duplex nPCR performed with the primers pol and LTR, the best primer concentrations were 7.5pM for the primer pol and 20pM for the LTR primers for both first and second rounds. In this step, there was a better result of duplex nPCR with an increase of dNTP concentration used to 200μM of each (Fig. 1).
, C−=negative Control, CA=CAEV standard sample, MV=standard MVV sample.")
Duplex nested-PCR. 1.3% agarose gel stained with ethidium bromide, presenting products of amplification of the pro-viral DNA target fragments of small ruminant lentiviruses samples. Products are equivalent to 1175bp for amplification with POL primers, 203bp for amplification with primers LTR and 187bp for amplification with primers GAG. MM=molecular marker (DNA ladder marker 100pb), C−=negative Control, CA=CAEV standard sample, MV=standard MVV sample.
To perform the agarose gel electrophoresis, different concentrations of agarose were tested. Among all the values tested, the 1.3% concentration allowed the best visualization of the bands. This concentration was adopted as the standard for the electrophoresis of samples obtained by duplex nPCR.
Evaluation technique performance in field samplesThe results obtained with the first duplex nPCR test with gag and pol primers using goat and sheep samples showed that pol primers were efficient for detection of large number of the positive samples of goats, but was not as efficient as the gag primers. From a total of 17 samples positive for gag, 15 showed positivity for pol and continued to demonstrate this expected result with the duplex technique (Fig. 2). In the evaluated sheep samples, only three were positive for the pol primers, demonstrating that the duplex technique performed with gag and pol primers is effective for more accurate detection of goat samples. All samples that were positive in the tests of individual nPCR for each set of primers also demonstrated positivity when tested with duplex nested PCR.
 stained with ethidium bromide, presenting products from the amplification of pro-viral DNA target fragments from small ruminant lentivirus samples. Products equivalent to 1175bp for amplification with pol primers and.")
The second step of standardization using the LTR and pol primers in field samples demonstrated that these primers are ideal for duplex nested-PCR, considering that the size of the bands formed is very different, allowing a good visualization of the results when analyzed on the agarose gel (Fig. 3). In addition, results demonstrated that LTR primers were more efficient at detecting sheep samples, failing to detect in samples from goats. The results demonstrated that pol primers were more specific for SRLV present in goats and LTR primers for SRLV obtained from sheep samples. These results also showed that the duplex nested-PCR technique is more sensitive to generate more reliable results for detection of lentivirus in sheep and goats.
 stained with ethidium bromide presenting products of the amplification of the pro-viral DNA target fragments of small ruminant lentivirus samples. Bands equivalent to 1175pb for amplification with POL primers and 203pb for amplification with primers LTR were observed. MM=molecular marker (DNA ladder marker 100pb), C−=negative control. Positive samples for pol –Goat 7 and Goat 8. Positive samples for LTR –Sheep 7, 10 and 15.")
Duplex nested-PCR. Agarose gel (1.3%) stained with ethidium bromide presenting products of the amplification of the pro-viral DNA target fragments of small ruminant lentivirus samples. Bands equivalent to 1175pb for amplification with POL primers and 203pb for amplification with primers LTR were observed. MM=molecular marker (DNA ladder marker 100pb), C−=negative control. Positive samples for pol –Goat 7 and Goat 8. Positive samples for LTR –Sheep 7, 10 and 15.
The analyses revealed that sequences from sheep and goats described here are related to genotype B1 (CAEV-Co) and genotype A (MVV K514) of SRLV, respectively. In addition, some genetic relationship was observed with other Brazilian strains described and the samples of sheep showed relation with Canadian strain (7-29Ont).
A phylogenetic tree (Fig. 4) was obtained using the NJ method with the gag nucleotide sequences, and this tree showed the relationship between the strains detected by duplex nPCR and the other known SRLV strains. The phylogenetic tree was determined using the neighbor joining (NJ) method35 implemented in MEGA, the Tamura-Nei gamma distance.36

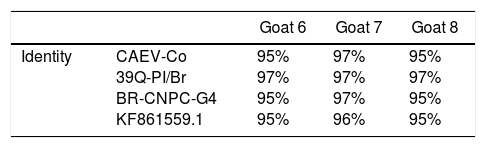
When comparing the sequences obtained with the samples deposited in the GenBank it was possible to verify the degree of identity between the strains. In Table 3 it is possible to verify the convergence and identity between the analyzed strains.
Similarity of the field strains with the strains used in the multiple sequencing and construction of the phylogenetic tree.
| Goat 6 | Goat 7 | Goat 8 | ||
|---|---|---|---|---|
| Identity | CAEV-Co 39Q-PI/Br BR-CNPC-G4 KF861559.1 | 95% 97% 95% 95% | 97% 97% 97% 96% | 95% 97% 95% 95% |
| Sheep 4 | Sheep 5 | Sheep 6 | ||
|---|---|---|---|---|
| Identity | MVV-K1514 7-29Ont Br/UFRGS-2/V27 21G-MG/Br | 93% 94% 94% 91% | 93% 94% 94% 91% | 93% 94% 94% 91% |
After analysis of the partial sequences of the gag gene from ovine and caprine samples the samples were submitted to the GenBank and are available for access with the following access numbers: MH251628 (Goat 6), MH251627 (Goat 7), MH251626 (Goat 8), MH251631 (Sheep 4), MH251630 (Sheep 5) and MH251629 (Sheep 6).
After the alignment of the samples under study with the standard samples, it is possible to identify the sites where differences in pro-viral DNA occur. These differences can be seen in Fig. 5.
Discussion
Several studies have shown that the molecular diagnosis of lentiviruses are increasingly sensitive,37–39 being a suitable technique for monitoring animals, particularly those of high zootechnical value. In this study, the developed technique proved to be efficient for the simultaneous detection of sheep and goat samples, allowing a faster and more sensitive diagnosis of lentiviruses compared to AGID. Considering that duplex nPCR allows the simultaneous diagnosis of different regions of the viral genome in a single assay, it is possible by means of a single assay to perform a more accurate diagnosis of the affected animals. In addition, costs of laboratory material and time are reduced when compared to the conventional nPCR technique.
The AGID technique is recommended by OIE12 and is widely used for the serological diagnosis of animals infected by SRLV. However, individual variations in immune response are observed and some animals may present low antibody titers, late seroconversion. In this study, among the 35 evaluated animals, comprised by sheep and goats, only five animals positive for SRLV infection in the AGID. Nevertheless, the proviral DNA of SRLV was detected in 27 animals with the nested-PCR. These findings demonstrate that the molecular technique was superior in detection capacity when compared to the AGID technique.
The results obtained corroborate the study carried out in breeding herds, in which animals known to be positive had negative results for AGID and positive for nPCR.40 In this context, several other studies have successfully demonstrated the use of PCR technique to detect pro-viral DNA of SRLV,15–18,37,39 considering that this technique presents high sensitivity collaborating for an early diagnosis.19
Barquero et al.10 consider that there is no “gold standard” test for the diagnosis of lentiviruses and with the advances in molecular biology several PCR protocols have been developed. Therefore, it is difficult to use the same primers in different geographic regions due to the SRLV genome heterogeneity.
In most studies involving CAEV41,42 and MVV37,43, this detection is performed using primers designed for the gag gene, since it has conserved sequences in different SRLV samples. However other regions are described that still have some degree of conservation and can be used to improve the sensitivity of the PCR technique, such as pol and LTR, allowing detection of a wide spectrum of CAEV and MVV field strains.10,44–46
The results of DNA sequence analysis demonstrated that the samples obtained from goats are close to the CAEV group, while the sheep samples are similar to those of MVV. In addition, through the analysis performed by the alignment of the sequenced samples, it was possible to verify the differences that occur in the viral genome, demonstrating the heterogenicity of the samples. Reinforcing the need for a more sensitive and specific test to detect a larger number of samples.
With the molecular tests, this study verified that the primers designed for the gag and pol regions presented high detection capacity for the circulating strains in the studied region with samples from goats. However, the primers of the gag region are more efficient. Gregory et al.,45 used pol primers to detect DNA pro-viral of CAEV in samples from lung, mammary gland, brain and synovial fluid, obtaining promising results.
For sheep samples in this study, gag and LTR primers were efficient in detecting positive animals. Several studies have described the use of PCR with primers LTR obtained good results.15,47 However, other researchers have shown that gag-PCR was more sensitive than LTR-PCR for detection of SRLV in sheep and goats.16,43,48
In our study, a similar detection was observed between the detection values of positive sheep samples using gag and LTR primers. However, one sample was positive for LTR and was negative for gag. This demonstrates that the primers for the LTR region are more specific for viral detection of sheep. This finding corroborates with the results obtained by Glaria et al.46 in which different tissue samples evaluated by gag-PCR and LTR-PCR were positive for LTR primers and negative for gag primers.
Similar results were obtained by Marinho et al.38 which reported that the detection of sheep lentiviruses was much more sensitive when performing nested-PCR with LTR primers than when using gag primers.
MVV infection was investigated in the same samples using two PCR tests directed to the LTR and pol sequences of the MVV gene. LTR-PCR was more sensitive and allowed to detect infection earlier than the other tests.49 Similar results were observed in the present study in which the samples of sheep were better detected with LTR-PCR than when the same technique was done using pol primers.
From these data, we can conclude that nested-PCR performed with primers from the pol region was not efficient for the detection of sheep samples in this study. However, when used for the detection of goat samples, it demonstrated high detection capacity. The nested-PCR performed with the gag primers was able to detect some sheep samples, but not all of them, which could lead to a false negative result when the PCR is performed only with the gag primers. Concerning primers for the LTR region, these have been shown to be the most efficient for the detection of sheep samples.
Considering the importance of diagnosing etiological agents involved in diseases to avoid the spread among animals, it is necessary to establish a system of rapid and highly specific diagnosis for these pathogens. A method that is sensitive, specific and capable of detecting all the variants of the virus even if present in low concentration are fundamental prerequisites for the success of the control and eradication of lentiviruses.3
In this sense, duplex nested-PCR technique for the diagnosis of lentiviruses of small ruminants allowed for a more accurate diagnosis, considering that it is an amplification reaction designed to detect multiple target sequences in the same sample. This promotes the detection of lentiviruses both in sheep and goats, even in the case of cross-infection. Through phylogenetic analysis, it can be concluded that the duplex nested-PCR technique was able to detect CAEV and MVV samples in a single assay.
In veterinary medicine, multiplex PCR has been developed and applied for the simultaneous detection of different diseases in pigs,50 cattle,51 birds,52 dogs53 and equines54 in a single assay.
As well as SRLV, equine herpesviruses are considered genetically and antigenically related, which promotes a difficulty in differentiating these pathogens through conventional serological tests.55 A multiplex nested-PCR technique was developed and successfully applied for the simultaneous detection of equine herpesviruses in different clinical samples. The same result could be obtained in this study with the application of a duplex nested-PCR technique.
The critical factors of PCR reaction are the selection of primers, the concentration of magnesium chloride and the annealing temperature.56 It was not necessary to change the magnesium concentration or the annealing temperature for performing the technique. However, for the duplex nested-PCR using pol and LTR primers, it was necessary adjustments in the mix to optimize the reaction. These alterations of the PCR mix for performing the duplex technique were also necessary in the study by Silva et al.57
The results obtained allowed us to conclude that the duplex nested-PCR is an important tool for a more accurate, sensitive and specific diagnosis of the different strains of SRLV that are circulating in Brazil, allowing a decrease of false negatives and increasing the capacity of viral detection.
Conflicts of interestThe authors declare no conflicts of interest.
This work was supported by the National Council of Scientific and Technological Development (CNPq), grant numbers 487425/2012-0 and 308995/2013-9; Coordination of Improvement of Higher Level Personnel (CAPES) for AUXPE-PROEX 533/2014 grant numbers 23038.003104/2014-58 and Cearense Foundation in Support of Scientific and Technological Development (FUNCAP), which provided a PhD scholarship. The authors are thankful to the Post Graduate Program in Veterinary Science (PPGCV) of the State University of Ceará for supporting the implementation of the experiments; to the Laboratory of Parasitic Diseases, particularly to Claudia Maria Leal Bevilaqua, PhD, for unconditional support in performing the molecular tests. The authors also express their gratitude to owners of the farms that allowed the use of their animals for this study.