






Serological testing and culling infected animals are key management practices aiming eradication of bovine leukemia virus infection. Here, we report the development of an indirect ELISA based on BLV recombinant capsid protein (BLVp24r) to detect anti-BLV antibodies in cattle serum. The BLVp24r was expressed in Escherichia coli and purified by affinity chromatography, and then used to set up the ELISA parameters. The Polysorp® plate coated with 50ng of antigen/well and bovine serum diluted 1:100 gave the best results during standardization. Using sera from infected and non-infected cattle we set up the cutoff point at 0.320 (OD450nm) with a sensitivity of 98.5% and specificity of 100.0%. Then, we tested 1.187 serum samples from dairy (736 samples) and beef cattle (451 samples) with unknown status to BLV. We found that 31.1% (229/736) and 9.5% (43/451) of samples amongst dairy and beef cattle, respectively, had IgGs to BLV. The rate of agreement with a commercial competitive ELISA was 84.3% with a κ value of 0.68. Thus, our BLVp24r iELISA is suitable to detect BLV infected animals and should be a useful tool to control BLV infection in cattle.
Bovine leukemia virus (BLV) is the etiological agent of a neglected, silent lifelong infection commonly found in dairy cattle1 named enzootic bovine leukosis (EBL). A large percentile of animal infected by the BLV might be asymptomatic or aleukemic (AL), at least at the initial stages of infection, while up to 30% develop a persistent lymphocytosis (PL)2 that in some cases, depending on the animal's Bola genotype,3 progresses to B cell lymphoma.4,5 The infection is considered eradicated in several parts of the world6 but is widely spread in North7–9 and most South American countries10 in which dairy farming is considered an important economic activity. There is no official data on the rate of infection in Brazilian dairy farm and most limited and biased epidemiological studies indicate that the range of seropositive animal varies from 12.5% to 100%.11,12 In South Brazil, in which most high productivity dairy farms are located, a within-herd infection rate usually exceeds 30%.13,14 BLV transmission might occur during pregnancy, calving or by ingesting colostrum from infected cows.15 In the herd, BLV spreading occurs mostly by management practices such as vaccination, dehorning, ear tagging, artificial insemination and uterine palpation8,16,17 that, when performed with the same equipment, might transfer BLV-infected cells from infected to non-infected animals.
Historically, BLV was considered a quite benign infection of cattle mainly because lymphomas were detected mostly in older animals that were still kept in the herd.1 However, in the last decade, several studies indicated that BLV negatively affects the productivity of dairy cows18–20 and that B and T lymphocytes from BLV-infected animals have impaired immune functions,21 which could affect the overall immune status by reducing the ability to respond to vaccine antigens22 and by predisposing to other infectious diseases including mastitis. Furthermore, a tremendous turnover on BLV research is on the way driven by data indicating that BLV might be associated with certain types of human cancer.23–25 Altogether, these data should encourage official measures to introduce compulsory diagnosis aiming to limit the spread of BLV and initiate control and eradication programs.
BLV infected animal produce a robust anti-BLV humoral immune response that might be detected by agar gel immunodiffusion (AGID) or by immune-enzymatic assays such as ELISA.26,27 In Brazil, antigen to AGID is produced on fetal lamb kidney (FLK) cells persistently infected with BLVs but, unfortunately, it is scarce and not reliable, and the use of overseas made ELISA kits for diagnosis is restricted by regulatory agency, discouraging even more voluntary diagnosis. Because several countries, mostly at the European Economic Community already eradicated EBL, the presence of BLV-infected animals in the herd might soon affect international trade of dairy products. Thus, controlling BLV spreading amongst dairy cattle by continuous diagnosis and culling infected animal should become mandatory to assure trading and safety of dairy products. With this in mind, in this study our major goal was to evaluate a recombinant BLV capsid protein as antigen to develop an indirect ELISA (iELISA) to diagnose BLV-infected cattle.
Material and methodsDNA extraction, cloning and sequencing of BLV capsid protein geneBlood samples from a cow infected by BLV (positive by AGID) was collected with EDTA and centrifuged (1300×g/10min) to obtain peripheral blood mononuclear cells (PBMC) from which total DNA was extracted using the Wizard®Genomic DNA Purification Kit (Promega, USA). The amount of DNA was measured by spectrophotometry and stored at −20°C until use.
PCR primers were designed to amplify a 666bp DNA fragment covering the complete nucleotide sequence of the BLV capsid protein. Two restriction sites were added in the primers to facilitate the directional cloning of the amplified fragment into the cloning vector (Forward (BamHI): 5′-ATTAGGGGATCCCCAATCATATCTGAAGGGAATCGCAA-3′; Reverse (HindIII): 5′-TGGCAGAAGCTTTTAGAGAAGTGCAGGCTGTTTCA-3′). The amplification was performed using 100ng of DNA, 0.4μM of each primer, 1.5U of Pfu DNA polymerase, 2.5μL of 10× buffer supplemented with MgSO4, 100μM of dNTPs (Promega, USA) and DNA/RNA-free water (Sigma, Brazil) to a final volume of 50μL. DNA was denatured at 95°C/5min, and then amplified by 35 cycles of denaturing (94°C/1min), annealing (52°C/40sec) and extension (72°C/2min) followed by a final amplification at 72°C for 10min. The resulting DNA fragment was analyzed by electrophoresis in agar gel (1%), purified and cloned into the pGEM-T-Easy vector (Promega, USA), and transformed into calcium competent TOP10 Escherichia coli (ThermoFisher Scientific®, USA). Penicillin-resistant clones were obtained and the presence of the capsid protein gene into the vector (pGEM-BLVp24) was confirmed by PCR and by sequencing using the vector forward primer (pUC/M13, Promega, USA). The sequence obtained was compared with reference genes available at GeneBank.
Expression of BLV capsid proteinThe pGEM-BLVp24 vector was digested with BamHI and HindIII (Promega, USA) restriction enzymes and analyzed by low melting point agar gel electrophoresis. The DNA fragment corresponding to the complete sequence that codifies de p24 capsid protein was cut out of the gel, purified (Wizard SV Gel, Promega, USA) and cloned into the pET-20 vector previously digested with the same restriction enzymes. The resulting plasmid (pET-20-BLVp24) was transformed into competent ER2566 E. coli (New England Biolabs, USA) for expression as a fusion protein containing histidine, a maltose binding protein (MBP) and a restriction site for the tobacco etch virus (TEV) protease at the N-terminus. The expression of the recombinant His-Mbp-TEV-BLVp24 (BLVp24r) was induced overnight with 0.1M isopropyl-β-D-thiogalactopyranoside (IPTG, Sigma, USA). The bacteria cells were pelleted (4000×g, 30min, 4°C), suspended in lysis buffer (20mM NaH2PO4, 500mM NaCl, 10mM Imidazol, pH 8.0) and sonicated thrice (70 watts) (Ultronic, Brazil). Then, the sonicated bacteria was centrifuged (13,000×g, 1h, 4°C) and the supernatant filtered (0.22μM) and purified using the Äkta Pure Chromatography System (GE Healthcare, Germany) connected to a HisTrap (GE Healthcare, Germany) column. The purified proteins were quantified, aliquoted and stored at −80° until use.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)Aliquots containing the BLVp24r protein eluted from the HisTrap affinity columns were analyzed by standard SDS-PAGE under reducing conditions (5% β-mercaptoethanol) using 5% acrylamide stacking gel overlaid on 10% acrylamide resolving gel. Samples collected prior to and after induction were also analyzed simultaneously. The SDS-PAGE gel was stained with Coomasie Blue R-250 (Bio-Rad Laboratories, USA) or used for Western blotting.
Production of polyclonal antibodies to the BLVp24rTwo Wistar rats were immunized twice (subcutaneous route, 21 days apart) with 50μg/dose of BLVp24r mixed to Montanide Gel 01 (15% v/v, Seppic, France). Blood samples were collected prior to and during immunization by puncturing the caudal vein, and final bleeding was performed by cardiac puncture. All procedures with rats were performed under anesthesia with Isoflurane (Cristalia, Brazil). The experimental protocol was approved by the local Committee on Animal Ethics and Usage (CEUA, protocol 12/2014).
Bovine serum samplesThe in house iELISA parameters were standardized using 100 bovine serum samples that were previously tested in our laboratory by AGID (Tecpar, PR). Amongst these samples 70 were negative and 30 positive to BLV antibodies. Out of the 70 negative samples, 16 were from cows that were also negative when tested by PCR targeting the Tax (nested PCR) and GAG BLV genes.25 A positive BLV reference serum (E 05) kindly provided by the OIE reference laboratory for EBL at the University of Leipzig (Germany) was also included on the study.
For the epidemiological study, we used 1.187 bovine serum samples from our Virology Diagnosis Laboratory and from the Virology Laboratory at the Federal University of Santa Maria (UFSM). Samples were obtained from dairy (n=736) and beef (n=451) cattle farms at the central region of Rio Grande do Sul, Brazil. Out of these, we randomly selected 255 serum samples and tested them also by a commercial anti-gp51 competitive BLV ELISA kit (Ingezim BLV Compac 2.0, Spain) to estimate the rate of agreement and Kappa (κ) values.
Western blottingAfter SDS-PAGE, BLVp24r were transferred to nitrocellulose membranes (Bio-Rad Laboratories, USA) using a semi-dry apparatus (Electrosystems, Brazil). The nitrocellulose membrane containing the proteins was blocked overnight at 4°C with phosphate buffered saline (PBS, pH 7.4) containing 0.05% Tween-20 (PBS-T) and 3% skim milk (PBS-TSK) under constant shaking and then cut into strip (4mm each). Serum collected from rats previously and after immunization with the BLVp24r, and serum from non-infected cows (IDGA/PCR negative; n=8) and cows naturally infected with BLV (n=8) were analyzed by western blotting. Rat or bovine serum samples were diluted 1:100 in 1% PBS-TSK and incubated with the membrane strips for 1h at 22°C under constant shaking. Then, the membranes were washed three times, 10min each, with PBS-T. Peroxidase conjugated anti-rat, or anti-bovine IgG whole molecule was diluted 1:1.000 and incubated with the membranes under the same conditions as the primary antibody. After washing thrice, the membranes were incubated with peroxidase substrate (4-Cloro-1-Naphthol+0.06% H2O2) for 10min and then transferred to distilled water to stop the reaction.
In house indirect ELISATwo commercially available polystyrene microplates (Maxisorp® and Polysorp®, Nunc, USA) were evaluated regarding the ability to adsorb the BLVp24r. The plates were coated with BLVp24r (2μg/well) diluted in carbonate buffer (pH 9.6) at 4°C for 12h and then washed three times with PBS-T. Plate wells were blocked with PBS-TSM at 37°C for 2h. Bovine serum positive (n=4) and negative (n=4) to BLV antibodies were diluted 1:100 in PBS-TSK 1%, added to the wells and allowed to react with the antigen for 1h at 37°C, in duplicates. After washing three times, peroxidase conjugated goat anti-bovine IgG (Sigma, USA) diluted 1:10.000 in PBS-TSK 1% was added and the plates were incubated as indicated above, followed by three washes and addition of substrate (3,3,5,5′-tetrametilbenzidina+0.06% H2O2). The plates were then incubated in the dark at 22°C for 10min and the reaction was stopped by adding 3N HCl. The plates were read at 450nm using a Synergy HI plate reader (BioTek®, USA). We used the optical density (OD) from three plates to calculate the mean positive/negative (P/N) ratio and the mean within-plate percent coefficient of variation (CV%) as previously described.28 The plates were then compared using an index obtained by dividing the P/N ratio by the CV%.
After selecting the best plate (Polysorp®), the optimal antigen concentration was evaluated in duplicates using different concentrations of the BLVp24r (2.0000, 1.000, 500, 250, 125, 100 and 50ng/well) in a final volume of 100μL, and sera from naturally infected (n=16) or non-infected cows (n=16). For this assay, the samples negative to BLV antibodies by the AGID were from cows that were also negative to BLV by PCR.25 The optimal antigen concentration was defined as the lowest antigen concentration that caused no significant changes in the absorbance obtained with the positive and negative sera.
The ideal serum dilution was determined by diluting bovine serum (negative and positive, n=16 each) from 1:25 to 1:200, and plates sensitized with the best antigen concentration determined in the previous step (50ng/well). The ideal dilution was determined taking into consideration the best specificity and sensitivity obtaining by the receiver operating characteristics (ROC) curve performed in all sera in duplicates.
The cut-off point was set by analyzing BLV negative (n=70) and positive (n=30) serum samples. Polysorp® plates were coated with BLVp24r (50ng/well) and serum samples were diluted 1:100. With the samples absorbance values, we determined the threshold following the ROC curve analysis.
Statistical analysisThe Kolmogorov–Smirnov test was used to determine the normal distribution of the data. The results were analyzed by Kruskal–Wallis or One Way Anova followed by Tukey post-test according to the data. Significant differences were considered when p<0.05. All the statistics were performed using the GraphPad Prism software (GraphPad, USA).
ResultsCloning, expression and characterization of BLVp24rA 666bp DNA fragment (Fig. 1A) was obtained and cloned into the pGEM-T-Easy vector and then sub-cloned into the pET20 aiming expression of a recombinant capsid protein from BLV (BLVp24r). The inserted fragment was sequenced and aligned to reference BLV genome (GenBank access number K02120, AP018032, LC080653, HE967302 and KT122858) resulting in 96–99% identity (data not shown).
 Polymerase chain reaction (PCR) amplification of the nucleotide sequence corresponding to the BLV capsid protein gene. Lymphocytes from BLV-infected cows (lanes 1) and from a non-infected cow (lane 2) were used for DNA extraction and analyzed by PCR. The fragment amplified was 666 nucleotides in length. M: 1 Kbp molecular weight markers. (B) Analyses of the recombinant BLV capsid protein (BLV p24r). Samples were collected prior to IPTG induction (lane 1), after overnight induction (lane 2), and after purification by the HisTrap column (lane 3), analyzed by SDS-PAGE and stained by Coomassie blue. The size of the molecular weight markers is indicated on the left side. (C) Western blot analysis of the BLVp24r protein. The purified BLVp24r protein resolved by SDS-PAGE and transferred to nitrocellulose membrane. Membrane strips containing the BLVp24r were incubated with serum from non-infected (panel C; n=8) and naturally BLV-infected (panel D; n=8) cows, and with rat serum (panel E) collected prior to (day 0) or after immunization (day 41) with the BLVp24r. All sera were also evaluated by our in house iELISA and the optical density (OD450nm) of each serum is indicated below the strips. M: molecular weight markers.")
(A) Polymerase chain reaction (PCR) amplification of the nucleotide sequence corresponding to the BLV capsid protein gene. Lymphocytes from BLV-infected cows (lanes 1) and from a non-infected cow (lane 2) were used for DNA extraction and analyzed by PCR. The fragment amplified was 666 nucleotides in length. M: 1 Kbp molecular weight markers. (B) Analyses of the recombinant BLV capsid protein (BLV p24r). Samples were collected prior to IPTG induction (lane 1), after overnight induction (lane 2), and after purification by the HisTrap column (lane 3), analyzed by SDS-PAGE and stained by Coomassie blue. The size of the molecular weight markers is indicated on the left side. (C) Western blot analysis of the BLVp24r protein. The purified BLVp24r protein resolved by SDS-PAGE and transferred to nitrocellulose membrane. Membrane strips containing the BLVp24r were incubated with serum from non-infected (panel C; n=8) and naturally BLV-infected (panel D; n=8) cows, and with rat serum (panel E) collected prior to (day 0) or after immunization (day 41) with the BLVp24r. All sera were also evaluated by our in house iELISA and the optical density (OD450nm) of each serum is indicated below the strips. M: molecular weight markers.
The expression of the pET-20-BLVp24 construction in E. coli ER2566 was analyzed by SDS-PAGE and the resulting fusion protein His-Mbp-TEV-BLVp24 (BLVp24r) had a molecular mass of 67kDa (BLVp24r+MBP 43kDa) (Fig. 1B). The expression yielded 14mg/L of ultra-pure BLVp24r.
The immunogenicity and reactivity of the BLVp24r were demonstrated by western blot using sera from immunized rats and sera from non-infected or from cows naturally infected with BLV. Sera from non-infected cows (Fig. 1C) and serum collected from rats prior to immunization failed to recognized BLVp24 (Fig. 1E-0) while sera from naturally infected cows and post-inoculation rat serum recognized the same protein (Fig. 1D and 1E-41). The reactivity of each serum used in the western blot was later evaluated by the standardized iELISA and the absorbance obtained is indicated in the lower panel.
MicroplatesThe mean OD obtained with negative and positive serum samples was significantly lower (p<0.01 and p<0.05, respectively) when tested with antigen adsorbed to Polysorp® microplate compared to the Maxisorp® microplate (not shown). However, the overall, analysis indicated a better efficiency with the Polysorp® microplates in that the P/N ratio obtained (4.5) was 2.2 times higher than the P/N ratio obtained with the Maxisorp® microplates (2.04). A lower CV was observed amongst negative and amongst positive samples with Polysorp® microplates which yielded a higher microplate index (Table 1). Considering that Polysorp® microplates allowed a better distinction between positive and negative samples (P/N ratio) compared to Maxisorp® microplates, they were then used on the remaining experiments.
Performance of Maxisorp® and Polysorp® ELISA microplates. The wells of both plates were coated with BLVp24r (2μg/well) and evaluated in triplicates with negative (n=4) and BLV-positive (n=4) sera samples to determine the P/N ratio (P/N) and the percentile of the coefficient of variation (%CV). The index value was then obtained by dividing the P/N ratio by the CV (%) from positive samples.
| Microplate | P/N | CV | Index | |
|---|---|---|---|---|
| Negative | Positive | |||
| Maxisorp | 2.04 | 7.1% | 8.8% | 0.23 |
| Polysorp | 4.50 | 2.2% | 4.1% | 1.1 |
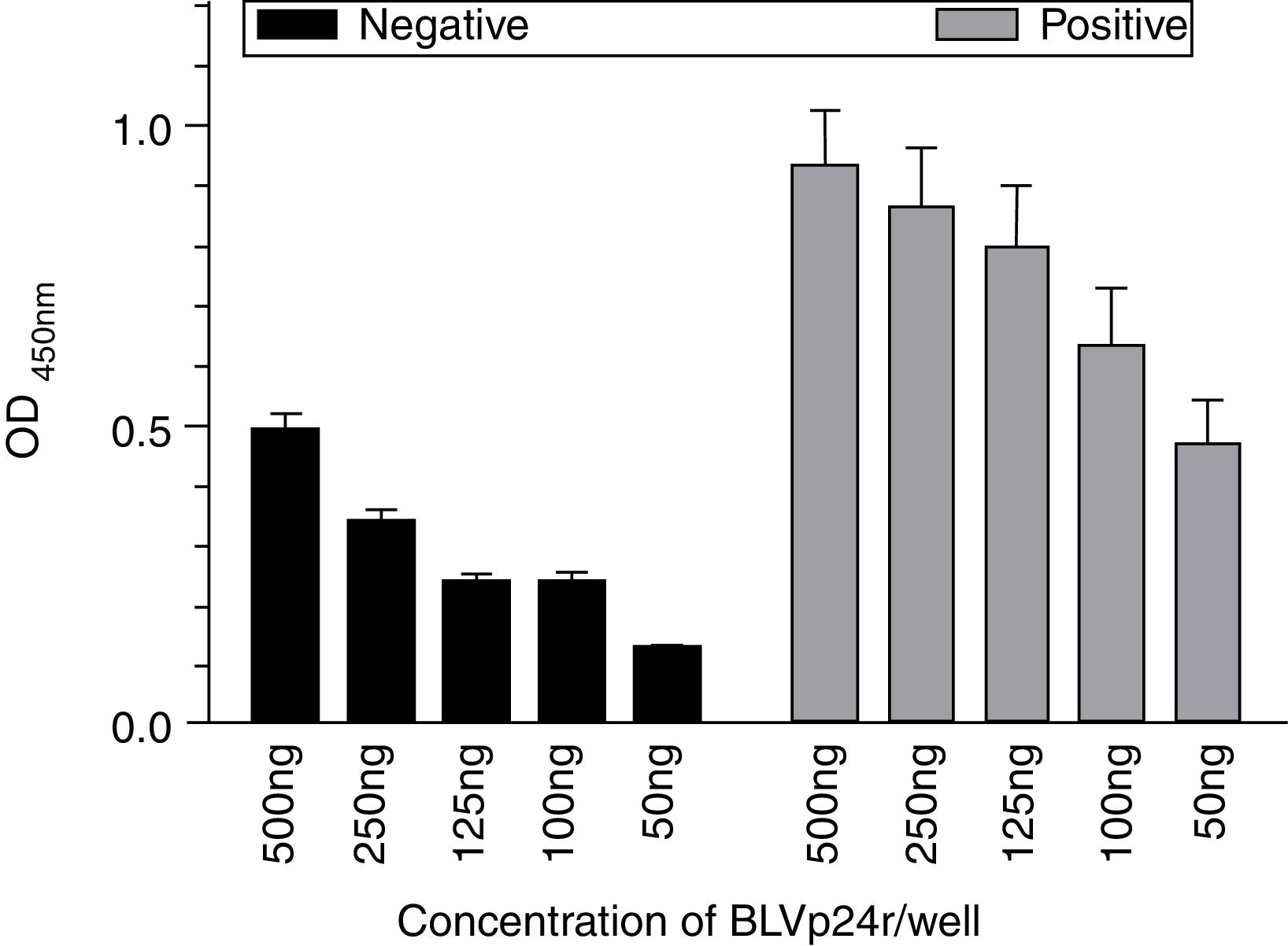
The ideal concentration of BLVp24r antigen to be used in the Polysorp® microplates was evaluated using a wide range of antigens dilutions and BLV negative (n=16) and positive (n=16) bovine serum. The mean serum absorbance reading obtained in each antigen dilution was not significantly different (p>0.05) from the next lower dilution and thus we choose the lower antigen concentration evaluated (50ng/well) to perform the remaining assay (Fig. 2).
 and naturally BLV-infected (n=16) cows diluted 1:100. The antigen concentrations evaluated are indicated. The results represent the OD mean±SEM at each antigen concentration. The arrow indicates the ideal antigen concentration.")
Ideal antigen concentration determination for the in house iELISA. Different antigen concentrations were adsorbed to the Polysorp® microplates and then incubated with serum from non-infected (n=16) and naturally BLV-infected (n=16) cows diluted 1:100. The antigen concentrations evaluated are indicated. The results represent the OD mean±SEM at each antigen concentration. The arrow indicates the ideal antigen concentration.
To set up the best serum dilution we tested samples diluted 1:25, 1:50, 1:100 and 1:200 with Polysorp® microplates coated with 50ng of BLVp24r/well. When all dilutions were compared, we observed a reduced sensitivity only with serum diluted 1:200 (83.33%) compared to 91.67% of sensitivity obtained with the other serum dilutions (not shown). Thus, we use the highest possible serum dilution (1:100) for the remaining studies.
Specificity, sensitivity and cut-off point determinationSeventy AGID negative sera samples and 30 AGID positive samples were used to determine the in house iELISA sensitivity and specificity for different cut-off point values. The ROC curve analysis of the in house iELISA data produced paired estimates of relative sensitivity and relative specificity at different cut-off values. A cut-off of OD≥0.320 was recommended. At this cut-off value we obtained a relative sensitivity of 98.5% (95% confidence interval=91.96–99.96%) and specificity of 100% (95% confidence interval=95.01–100%) with a likelihood ratio of 75. The ROC curve had an area under the curve (AUC) value of 0.9989 (95% confidence interval=0.9961–1.002; p<0.0001) demonstrating a high level of accuracy for this iELISA (Fig. 3). Thus, samples with an absorbance higher than 0.320 were considered positive when the negative control was at least 1.5 times smaller than the cut-off value.
 analysis. The ROC curve was created using the results obtained from analyzing 70 BLV negative and 30 BLV positive bovine serum samples (diluted 1:100) by the in house iELISA (50ng antigen/well) performed with the Polysorp® microplate. The area under the ROC curve was 0.9989.")
Receiver operating characteristic (ROC) analysis. The ROC curve was created using the results obtained from analyzing 70 BLV negative and 30 BLV positive bovine serum samples (diluted 1:100) by the in house iELISA (50ng antigen/well) performed with the Polysorp® microplate. The area under the ROC curve was 0.9989.
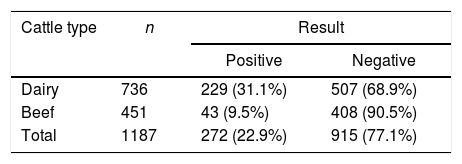
By using our in house iELISA, we analyzed 1.187 sera samples from dairy (736) and beef (451) cattle from the North region of Rio Grande do Sul State. Within dairy cattle, we found 229 (31.1%) positive samples and within beef cattle only 43 (9.5%) samples had antibodies to BLVp24r (OD>0.320) (Table 2).
Anti-BLV antibodies prevalence in dairy and beef cattle. Serum samples from dairy cattle (n=736) and beef cattle (n=451) were evaluated in duplicates by the in house iELISA, using 50ng/well of BLVp24r in Polysorp® microplates and a 1:100 sera dilution. Samples with a OD>0.320 were considered positive.
| Cattle type | n | Result | |
|---|---|---|---|
| Positive | Negative | ||
| Dairy | 736 | 229 (31.1%) | 507 (68.9%) |
| Beef | 451 | 43 (9.5%) | 408 (90.5%) |
| Total | 1187 | 272 (22.9%) | 915 (77.1%) |
The performance of the in house iELISA to detect anti-BLV antibodies was compared with the commercial anti-gp51 competitive ELISA assay. We randomly selected 255 sera previously tested in the in house iELISA (118 negative and 137 positive) and evaluated them in duplicates using the commercial ELISA. We found 122 negative and 133 positive samples (Table 3) resulting in 84.3% of agreement and a Kappa (K) index of 0.68 (good agreement).
Rate of agreement between the iELISA (anti-p24 antibodies) and a commercial competitive ELISA (anti-gp51 antibodies) evaluated using 255 samples selected randomly amongst dairy cattle serum samples.
| Competitive gp51 ELISA | ||||
|---|---|---|---|---|
| Positive | Negative | Total | ||
| iELISA BLVp24r | Positive | 115 | 22 | 137 |
| Negative | 18 | 100 | 118 | |
| Total | 133 | 122 | 255 | |
Enzootic bovine leukosis usually occurs as a silent, asymptomatic disease in most dairy farms and for this reason it has been neglected for decades. Up to 30% of infected animal develop a persistent lymphocytosis (PL) which in some animal might progress to B cell lymphoma. There are no vaccines and all infected animals respond to infection by producing antibodies firstly to the gp51 viral envelope glycoprotein and then to the p24 viral capsid protein. These viral antigens have been globally explored for serological diagnosis of BLV infection; the gp51 is used mostly, but no exclusively, in AGID assays and the p24 in immunoassays such as iELISA.26,29–33 Infected animals might also be detected by PCR targeting conserved viral genes33; although highly sensitive, PCR usage for epidemiological studies is not practical and convenient. In Brazil, the AGID assay is the only official diagnostic method accepted but, unfortunately, antigen production is limited and the assay yields unreliable results that frustrates diagnosticians and difficults a rational approach to disease control. In addition, because the possible involvement of BLV with certain types of human cancer,23,24 EBL control in dairy cattle becomes a matter of public health and should become mandatory.
With this in mind, here we describe the production of a recombinant BLV capsid protein (BLVp24r) and its use for development of an indirect ELISA assay to detect anti-BLV antibodies in blood samples. The vector and expression system was chosen aiming to improve the recombinant protein yield, solubility and purification without losing immunogenicity and recognition by antibodies from BLV naturally infected animals. Indeed, the recombinant protein was easily purified by standard procedures and its immunogenicity demonstrated in rats. Furthermore, the protein was recognized by antibodies from BLV naturally infected cows, both by western blot and ELISA, and by the OIE reference serum E 05, kindly provided by the OIE reference laboratory for EBL at the University of Leipzig (Germany), that was also used in the iELISA settings. In addition, the MBP tail is not immunogenic in rats, or even other species, which allows the recombinant fusion protein to be used in developing diagnostic assays.34
We found that microplate characteristics affected the outcome of the assay. Overall, negative and positive sera samples tested with the Polysorp® microplate had a lower absorbance reading but the P/N ratio and the %CV found with both types of plates indicated that the Polysorp® microplates should be used to better differentiate negative from positive samples. Interestingly, in a previous study in which we expressed the capsid protein of Hepatitis E Virus (HEV) in the same expression system we found that Maxisorp® microplates had a higher antigen binding efficiency and a lower CV compared to Polysorp® microplates.28 We are aware that antigen adsorption on the solid phase can be affected by hydrophobic and hydrophilic domains present on the molecules and by the type of plate, underlying the necessity to test different plates during the initial step of an ELISA optimization for a target antigen. Considering the maximal binding capacity/cm2 of the well (0.5μg protein) and the final volume used in the assay (100μL), we estimated that saturation would be achieved with 0.37μg of antigen. However, the differences on P/N ratio and %CV were not significant when we compared different antigen concentration and, therefore, we choose the lowest antigen concentration tested (50ng/well) to carry out the study. Even though antigen concentration might affect ELISA parameters, similar recombinant antigen concentrations were used in previous study to detect BLV30 and other viral infections.35 Furthermore, our BLV p24r was fused to MBP and resulted in a protein with a higher molecular mass (67kDa) when compared to the BLV capsid protein per se (24kDa). When larger proteins are used, lower concentrations (μg/well) might be required to optimize ELISA parameters. We hypothesize that MBP would contribute to protein adsorption to the microplate well leaving relevant antigenic epitopes completely uncovered and available improving the efficiency of antibody binding to the recombinant protein and reducing the amount of antigens required to optimize ELISA parameters.
The performance of our in house iELISA was compared to the AGID and a commercial competitive ELISA that used a peroxidase-labeled monoclonal antibody as detecting antibody. There was a 72% agreement between the in house iELISA and AGID and the kappa index was low (0.42; data not shown). However, in our experience with the available AGID assay, we noticed that even the positive control serum provided with the kit commonly fails to give a reliable result, which is difficult to read even by experienced diagnosticians. Furthermore, because iELISA is more sensitive than AGID in detecting even small amounts of antibody to a given antigen,27,32 animals recently infected might not be detected by AGID. However, when compared to the commercial ELISA, we found 84.3% of agreement between the results with a κ value of 0.68 and the predictive positive and negative values were 0.839 and 0.847, respectively. Although the agreement found between the assays is considered good, it is worth nothing that the commercial assay detects antibodies to the BLV gp51 glycoprotein using a peroxidase-labeled competing monoclonal antibody. Thus, in this case, because gp51 is a highly variable surface glycoprotein, animals infected with a different genotype10 might not have antibodies to the epitope targeted by the detecting monoclonal antibody and would result negative in the commercial but positive in the in house iELISA that uses the highly conserved BLV p24r protein. In fact, one animal positive by iELISA and Western Blot assay (Fig. 1C, second strip OD450 0.997) ended negative when assayed by the commercial ELISA, strengthening the hypothesis that different genotypes of BLV are circulating on different geographical areas and would escape detection by serological assay that would target unique epitopes. In the other hand, animals negative to the in house iELISA but positive in the commercial assay could have been infected recently and developed antibodies mostly to the viral surface glycoprotein. Thus, in this situation, the ideal iELISA should contain both p24 and gp51 antigens no minimize the possibility of false-negative results. In any case, controlling BLV infection in a herd requires periodical testing to remove positive animals as long as any positive animal is still detected within the herd.
BLV causes a silent, life-long infection of dairy cattle which has been mostly underestimated or even overshadowed by other diseases. However, as the economic impact of BLV on cow's health and milk production is appreciated18–20,36,37 and, most recently, with its possible link with human breast cancer,23,25 a major switch on diagnosis and disease control should be expected. Thus, rapid and reliable serological test should be promptly available at low cost to milk farmers and eradication policies should become mandatory. Using our in house iELISA, we tested 736 dairy and 451 beef cattle sera samples from the North region of Rio Grande do Sul and found a prevalence of 31.1% (229/736) and 9.5% (43/451) respectively. For dairy cattle, the prevalence to BLV infection remains similar to that reported in previous studies.13,14 In fact, we were expecting a higher prevalence. Thus, we hypothesize that current invasive management procedures like ear tagging, vaccination, artificial insemination and tuberculosis testing are performed taking into consideration the risk of transmitting BLV amongst cattle, mainly because most veterinarians are aware of the relatively high prevalence of within herd infection already reported in dairy cattle from this region. By doing so, iatrogenic transmission of BLV is kept at minimum. Also, we are aware that most veterinarians recommend that once an infected animal is detected, it should be removed from the herd or managed in such a way to reduce the risk of transmitting BLV to non-infected cows.
In conclusion, the in house iELISA we developed using BLVp24r might be explored as a commercial test to detect BLV infected animals. The protein expressions and purification system we used yields enough protein to make it affordable and simple to use. Because BLV is widespread in dairy cattle and we showed a high rate of infection also in beef cattle, and because of the recent reports of BLV infection in humans, controlling BLV infection in cattle becomes a public health issue that should become mandatory.
Conflicts of interestThe authors declare no conflicts of interest.
Ana Paula Andreolla was a Master Student supported by the Fundação Universidade de Passo Fundo and then by CAPES (01947247000). L.M.S. Erpeen has an undergraduate fellowship from the Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq. The Laboratório de Microbiologia e Imunologia Avançada is equipped with funding from the Secretaria de Desenvolvimento Econômico, Ciência e Tecnologia (SDECT – Convênio 46/2014, processo 328-25.00/14-0). L.C. Kreutz holds a fellowship from CNPq (Produtividade em Pesquisa – PQ 307900/2016-9).