


A modified TaqMan real-time polymerase chain reaction targeting a 138bp fragment within the lipl32 gene was developed to identify exclusively pathogenic Leptospira spp. in dog urine samples. Thirty-five samples from dogs with suspected clinical leptospirosis and 116 samples from apparently healthy dogs were tested for presence of leptospiral DNA using the TaqMan-based assay. The results were compared with those from a well-established conventional PCR targeting the 16S RNA encoding gene associated with nucleotide sequencing analysis. The overall agreement between the assays was 94.8% (confidence interval [CI] 95% 88–100%). The newly developed assay presented 91.6% (CI 95% 71.5–98.5%) relative sensitivity (22[+] lipl32 PCR/24[+] 16S RNA and sequencing), 100% (CI 95% 96.3–100%) relative specificity and 98.7% accuracy (CI 95% 94.8–100%). The lipl32 assay was able to detect and quantify at least 10 genome equivalents/reaction. DNA extracted from 17 pathogenic Leptospira spp., 8 intermediate/saprophytic strains and 21 different pathogenic microorganisms were also tested using the lipl32 assay, resulting in amplification exclusively for pathogenic leptospiral strains. The results also demonstrated high intra and inter-assay reproducibility (coefficient of variation 1.50 and 1.12, respectively), thereby qualifying the newly developed assay as a highly sensitive, specific and reliable diagnostic tool for leptospiral infection in dogs using urine specimens.
Leptospirosis is a reemerging bacterial disease of global importance caused by pathogenic spirochetes of the genus Leptospira.1 Pathogenic Leptospira is currently classified into 10 distinct genomospecies and more than 260 serovars,2 which can affect a wide variety of mammalian species, including dogs and humans. Leptospiral transmission occurs through direct contact with contaminated biological material or indirectly through contact with contaminated environmental water resources.3 Thus, identification of infected animals is a key strategy for preventing the disease.
Canine leptospirosis is frequently reported worldwide1,4 and infected dogs manifest a broad spectrum of clinical symptoms, ranging from hepatic and renal failure, often accompanied by hemorrhagic and pulmonary disorders, to mild, self-limiting febrile illness.3 Although susceptible to many serovars, dogs can act as maintenance hosts for different pathogenic Leptospira spp., notably L. interrogans sv. Canicola, thus contributing to the spread of leptospires into the environment without overt clinical signs of infection.1
The diagnostic approach for canine leptospirosis is predominantly based on serological evidence of infection, usually obtained through the microscopic agglutination test (MAT).5 However, use of MAT presents several limitations and the serological results should be interpreted with parsimony: MAT presents low sensitivity during the early stages of the disease, and multiple samples from both the acute and the convalescent phases are usually required for confirming the infection.6,7 Additionally, serum titers may therefore represent recent exposure to the pathogen instead of active infection,6,7 and the interpretation of serological data can be particularly confusing in relation to recently immunized dogs or those with unknown immunization status. The identification of chronically infected individuals using MAT can also be challenging, given that serum titers may not necessarily be associated with asymptomatic renal carriage of leptospires.3
Although culturing leptospires is still considered the eligible test to unmistakably confirm leptospiral infection, recovery of viable leptospires from culture media faces critical drawbacks, such as frequent contamination, fastidious growth of the pathogen and low diagnostic sensitivity,1 which undermine its use in clinical and epidemiological studies. On the other hand, PCR has been successfully used to confirm leptospiral infection and enables early diagnosis of clinical leptospirosis,8,9 along with identification of dogs with asymptomatic urinary shedding of leptospires.10–13
A great number of PCR assays targeting different protein-encoding genes have been developed for diagnosing human and animal leptospirosis.5 Different quantitative PCR protocols targeting the lipl32 gene have previously been described11,14–16; however, few studies have validated these assays for detecting leptospires in canine specimens.11,14 LipL32 is an abundant outer membrane protein that occurs exclusively in pathogenic Leptospira and presents highly pairwise DNA sequence identity among pathogenic species.17
The current report describes the development and validation of a modified quantitative PCR assay targeting the lipl32 gene for identifying pathogenic Leptospira in canine urine samples.
Material and methodsSamples and isolatesUrine samples from 35 dogs with suspected clinical leptospirosis that were attended at the University of São Paulo Veterinary Hospital Service (Hovet FMVZ-USP) between 2013 and 2015 were used to evaluate the diagnostic sensitivity, specificity and accuracy of the TaqMan PCR assay. The dogs included in this study were considered suspected for acute leptospirosis when presenting high blood urea nitrogen (BUN) and creatinine levels (above 60mg/dL and 1.4g/dL, respectively), in association with two or more typical clinical manifestations of leptospirosis (hemorrhagic disorders, fever, jaundice, prostration or muscle weakness). An additional 116 urine samples from apparently healthy dogs that were being kept in two different local public shelters, which were collected during 2013, were also tested.
The Ethics Committee of the School of Veterinary Medicine and Animal Science, University of São Paulo (protocols 2706/2012 and 2406140614) approved all procedures involving animal manipulation in the current study.
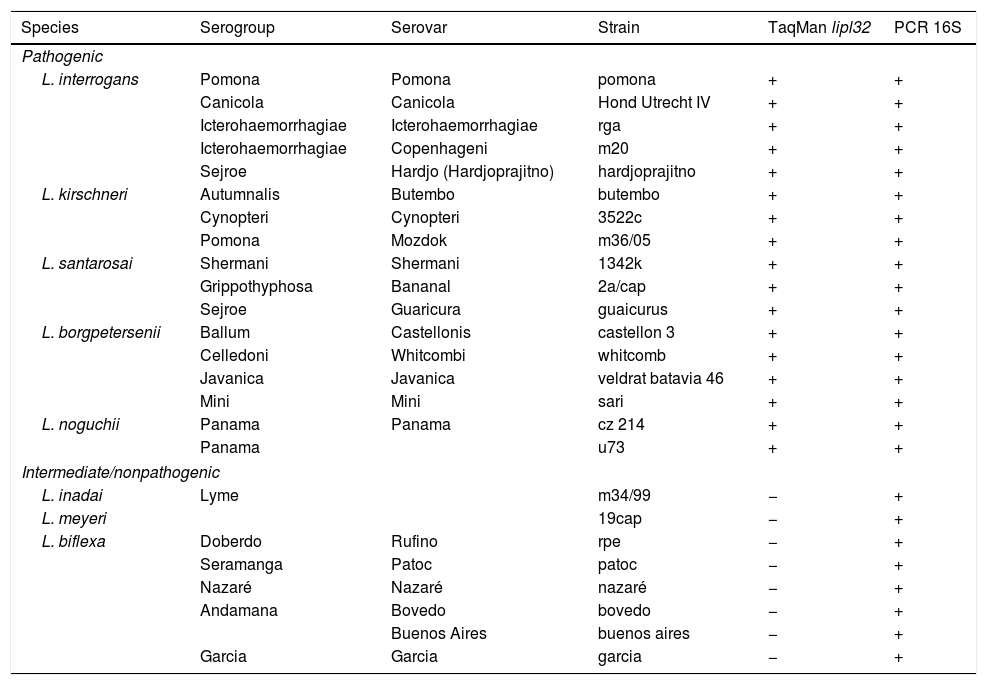
The leptospiral strains used in this study were provided by the Bacterial Zoonosis Laboratory, Department of Preventive Veterinary Medicine and Animal Health, School of Veterinary Medicine and Animal Science, University of São Paulo. The isolates (Table 1) were maintained in semisolid Fletcher medium (BD Diagnostics, Sparks, MD, USA) containing 1.5% agar (BD Diagnostics) at 28°C under aerobic conditions, and were collected at mid-log phase.
Pathogenic, intermediate and nonpathogenic Leptospira strains used for sensitivity and specificity tests and results from TaqMan real-time PCR and 16S PCR assays.
| Species | Serogroup | Serovar | Strain | TaqMan lipl32 | PCR 16S |
|---|---|---|---|---|---|
| Pathogenic | |||||
| L. interrogans | Pomona | Pomona | pomona | + | + |
| Canicola | Canicola | Hond Utrecht IV | + | + | |
| Icterohaemorrhagiae | Icterohaemorrhagiae | rga | + | + | |
| Icterohaemorrhagiae | Copenhageni | m20 | + | + | |
| Sejroe | Hardjo (Hardjoprajitno) | hardjoprajitno | + | + | |
| L. kirschneri | Autumnalis | Butembo | butembo | + | + |
| Cynopteri | Cynopteri | 3522c | + | + | |
| Pomona | Mozdok | m36/05 | + | + | |
| L. santarosai | Shermani | Shermani | 1342k | + | + |
| Grippothyphosa | Bananal | 2a/cap | + | + | |
| Sejroe | Guaricura | guaicurus | + | + | |
| L. borgpetersenii | Ballum | Castellonis | castellon 3 | + | + |
| Celledoni | Whitcombi | whitcomb | + | + | |
| Javanica | Javanica | veldrat batavia 46 | + | + | |
| Mini | Mini | sari | + | + | |
| L. noguchii | Panama | Panama | cz 214 | + | + |
| Panama | u73 | + | + | ||
| Intermediate/nonpathogenic | |||||
| L. inadai | Lyme | m34/99 | − | + | |
| L. meyeri | 19cap | − | + | ||
| L. biflexa | Doberdo | Rufino | rpe | − | + |
| Seramanga | Patoc | patoc | − | + | |
| Nazaré | Nazaré | nazaré | − | + | |
| Andamana | Bovedo | bovedo | − | + | |
| Buenos Aires | buenos aires | − | + | ||
| Garcia | Garcia | garcia | − | + | |
Urine samples were collected via urinary catheterization or cystocentesis and were centrifuged within 2h after collection (6500×g for 25min at 25°C) as previously described.16,18 The pellets were resuspended in 2mL of sterile phosphate-buffered saline (PBS; 0.01M PO4; 0.1M NaCl; pH 7.2) and all DNA extractions were processed not more than 48h after collection. In order to access the performance of the DNA extraction procedures and verify the presence of PCR inhibitors, all clinical samples were subjected to quantitative amplification targeting the melanocortin 1 receptor-encoding gene (MC1R), a nuclear gene that encodes for a seven-pass transmembrane G protein-coupled receptor protein that is involved in hair and fur coloring in mammals.19 Genomic DNA from clinical samples was extracted using NucliSens® miniMAG™ (BioMérieux Inc., Durham, NC, USA), in accordance with the manufacturer's instructions, with slight modifications: 1mL of resuspended solution was used in the initial lysis step and the final elution step was performed with 40μL.
Additionally, pure cultures of leptospires and non-spirochetal pathogens used to determine the analytical specificity of the lipl32 TaqMan real-time PCR were subjected to two washing steps (6500×g, 25min at 25°C) using sterile PBS prior to DNA extraction; the pellets were resuspended in 2mL of sterile phosphate-buffered saline and genomic DNA was extracted using QIAamp DNA Mini Kit® (Qiagen Inc., Valencia, CA), in accordance with the manufacturer's instructions.
Primer and probe designPrimers and probe targeting the lipl32 gene were selected based on a previously reported real-time PCR reaction described by Rojas et al.11 A total of 113 sequences encoding the LipL32 protein that were available in the National Center for Biotechnology Information Nucleotide Database (NCBI; http://www.ncbi.nlm.nih.gov/) were aligned using the Mega 5.10 software20 in order to evaluate the in silico specificity of the previously described primers. The selected sequences included all available L. interrogans sv. Canicola sequences, and sequences from L. interrogans serovars that are commonly attributed to canine leptospirosis (serovars Icterohaemorrhagiae, Pomona and Grippotyphosa), along with other representative serovars of the species L. interrogans, L. kirschneri, L. borgpetersenii, L. noguchii, L. weilii and L. santarosai (Supplementary Material 1). Two of the sequences thus selected (AY461924 and DQ286416) presented one nucleotide mismatch with the degenerated forward primer previously described.11 Oligonucleotide structural analysis of the described primers was performed using OligoAnalyser 3.1 (http://www.idtdna.com/), which showed the possibility of self-dimer and hairpin formation. To accommodate all available sequences tested and to overcome possible structural primer instability, a newly non-degenerated forward primer was designed (5′-TAAAGCCAGGACAAGCGCC-3′), resulting a 138bp amplicon when combined with the reverse primer previously described, which was not modified. The fluorescent probe sequence described by Rojas et al.11 was also not modified, except for use of minor groove binder (MGB) as a quencher at the 3′-end instead of nonfluorescent quencher (NFQ).
TaqMan real-time PCR assaysOptimal concentrations of primers (100, 300, 500, 600, 700 and 900nM) and probe (200, 250 and 300nM) targeting the lipl32 gene were tested in accordance with the manufacturer's recommendations (Applied Biosystems, Thermo Fisher Scientific Inc., Carlsbad, CA, USA). This resulted in an optimized reaction with a final volume of 25μL using 600nM of each primer, 250nM of the probe, 1× TaqMan® Universal Master Mix II (Thermo Fisher, Scientific Inc, Carlsbad, CA, USA), DNase-free water and 5μL of extracted DNA. The amplification protocol consisted of 2min at 50°C, 10min at 95°C and 45 cycles of amplification (95°C for 15s and 60°C for 60s).
The MC1R assay was carried out using primers and probe previously described by Kanthaswamy et al.21 The reaction consisted of 1× TaqMan® Universal Master Mix II (Thermo Fisher, Scientific Inc, Carlsbad, CA, USA), 900nM of each primer, 250nM of the probe, sterile DNase-free water and 5μL of extracted DNA, resulting in a final volume reaction of 25μL. The cycling conditions consisted of an initial step of 50°C for 2min, followed by 10min at 95°C and 40 cycles of amplification (95°C for 15s and 60°C for 60s). All MC1R runs were performed concomitantly with analysis on DNA extracted from pure cultures of canine fibroblasts, as positive controls.
All TaqMan real-time PCR runs were performed in the same machine (Applied Biosystems® 7500 Real-Time PCR System, Thermo Fisher Scientific Inc, Carlsbad, CA, USA).
Analytical sensitivity of the lipl32 assayThe analytical sensitivity of the lipl32 assay was determined through amplification of serial dilutions of genomic DNA extracted from a pure culture of L. interrogans serovar Canicola strain Hond Utrecht IV. DNA was extracted from a 2mL sample and quantified in duplicate using a Qubit® 2 fluorimeter (Invitrogen, Thermo Fisher Scientific Inc, Carlsbad, CA, USA) with a Quant-iT™ DNA Assay Kit (Invitrogen, Thermo Fisher Scientific Inc, Carlsbad, CA, USA). The number of Genomic Equivalents (GEs), assuming one genome copy per leptospire, was estimated based on a genomic size of 4.659Mb. To prepare standard curves, the extracted DNA was standardized to an initial concentration of 1×106GE/reaction, in order to perform serial 10-fold dilutions until reaching 1×101GE/reaction. All standard-curve dilutions were tested in triplicate, and each run included a single negative control containing sterile nuclease-free water. The lower limit of detection (LLOD) was determined as the concentration at which 95% or more of the replicates from eight runs (performed on different occasions) presented amplification. The cycle threshold (Ct) cutoff value was determined by calculating the average Ct values of LLOD replicates from different standard-curve runs. Coefficients of variation (CV) and average Ct values were calculated to define inter and intra-assay variations and estimate the assay reliability. The CV values were calculated using the standard deviation (SD) and the mean Ct using the formula CV=(SD×100%)/mean Ct.
Analytical specificity of the lipl32 assayThe specificity of the new set of primers targeting the lipl32 gene was initially confirmed by BLAST analysis (http://blast.ncbi.nlm.nih.gov/), followed by performing the assay using extracted DNA from several spirochetes as a template, including pathogenic strains, saprophytic strains and strains with intermediate pathogenicity. DNA extracted from pure cultures of other non-leptospiral pathogens was also tested, including Brucella canis (strain RM6/66 – ATCC 23365), Enterococcus faecalis (field strain), Escherichia coli (strain DH5α), Salmonella enterica sv. Typhi (ATCC 14028), Yersinia enterocolitica (field strain), Proteus mirabilis (field strain), Candida albicans (ATCC 90112), Staphylococcus epidermidis (field strain), Staphylococcus aureus (ATCC 25923), Streptococcus pyogenes (field strain), Klebsiella pneumoniae (field strain), Pseudomonas aeruginosa (ATCC 27853), Serratia marcescens (field strain), Mycobacterium tuberculosis (strain H37Rv), Mycobacterium bovis (strain AN5), Rickettsia rickettsii (strain TAIAÇU), Rickettsia bellii (strain MOGI), Rickettsia parkeri (strain AT24), Ehrlichia canis (strain SÃO PAULO), Leishmania infantum chagasi (strainCBT56 RIO GRANDE DO NORTE) and Toxoplasma gondii (strain RH).
Diagnostic sensitivity, specificity and accuracy of the lipl32 assayQuantitative amplification of DNA extracted from clinical samples that were obtained from symptomatic and asymptomatic dogs was performed in triplicate. The samples were considered positive when at least two thirds of the replicates presented Ct values lower than the cutoff value that had been established. For comparative analysis, the performance of the modified lipl32 TaqMan real-time PCR assay was compared with results obtained from a well-established conventional PCR in which a Leptospira genus-specific set of primers targeting a 331bp fragment of the 16S rRNA gene (Lep1 F and Lep2 R) was used.22 Conventional PCR amplification was carried out as follows: 94°C for 5min, 40 cycles at 94°C for 30s, 60°C for 30s, 72°C for 30s and a final extension at 72°C for 5min. Pure L. interrogans serovar Canicola (strain Hond Utrecht IV) genomic DNA was used as a positive control and DNase-free water as a negative control in all PCR runs. The amplified products were separated by electrophoresis on a 2% agarose gel stained with SYBR Safe DNA gel stain (Invitrogen, Thermo Fisher Scientific Inc, Carlsbad, CA, USA) and were analyzed under UV transillumination. The nucleotide sequences of the amplified fragments were sequenced using the BigDye Terminator 3.1 cycle sequencing kit (Thermo Fisher Scientific Inc, Carlsbad, CA, USA) on an ABI 7500 Genetic Analyzer (Thermo Fisher Scientific Inc, Carlsbad, CA, USA), in accordance with the manufacturer's recommendations. The sequences obtained were compared with those in the GenBank database (http://www.ncbi.nlm.nih.gov/BLAST), to determine species identification. Agreement between the assays was assessed using the kappa test, with a 95% confidence interval, along with calculation of relative sensitivity, specificity and accuracy. The 95% confidence intervals (CI) for the sensitivity and specificity values were calculated as proportion CI. The 95% confidence intervals for proportions (sensitivity, specificity and accuracy) were calculated, as all the other statistics, using the software RStudio (Version 1.0.136 – © 2009–2016 RStudio, Inc).
ResultsThe modified lipl32 TaqMan real-time PCR assay was able to detect 1×106 to 1×101 GE/reaction of the extracted leptospiral DNA in a linear manner, with a coefficient of correlation (of 0.998 and efficiency of 98.96%. The analytical data also showed that the assay was able to detect 10 copies per reaction (LLOD): from 42 replicates containing 10 copies/reaction from eight different runs, 41 replicates presented amplification.
The reaction presented a Ct cutoff value of 37.5 arbitrary units (SD ± 0.324) and was determined based on the average Ct value of 17 replicates amplifying 10 copies per reaction from five different standard-curve runs. The standard curves were selected based on adequate efficiency values (ranging from 92.74% to 98.96%) and high Ct values (> 38) were not considered in the calculation to avoid unspecific amplifications. All selected runs were performed on separate days and the highest intra-assay and inter-assay CV values considering all standard-curve dilutions were 1.50 and 1.12, respectively (Supplementary Material 2).
The lipl32 TaqMan real-time PCR assay was able to detect DNA from all pathogenic Leptospira spp. tested, while none of the intermediate or saprophytic Leptospira strains presented amplification (Table 1). All strains tested were also subjected to 16S rRNA amplification to confirm the appropriate extraction procedure, and all samples yielded strong positive bands when analyzed on agarose gel (Table 1). Moreover, the new set of primers used did not show any unspecific hybridization in the in silico evaluation and no amplification was observed when DNA from other non-spirochetal pathogens was tested.
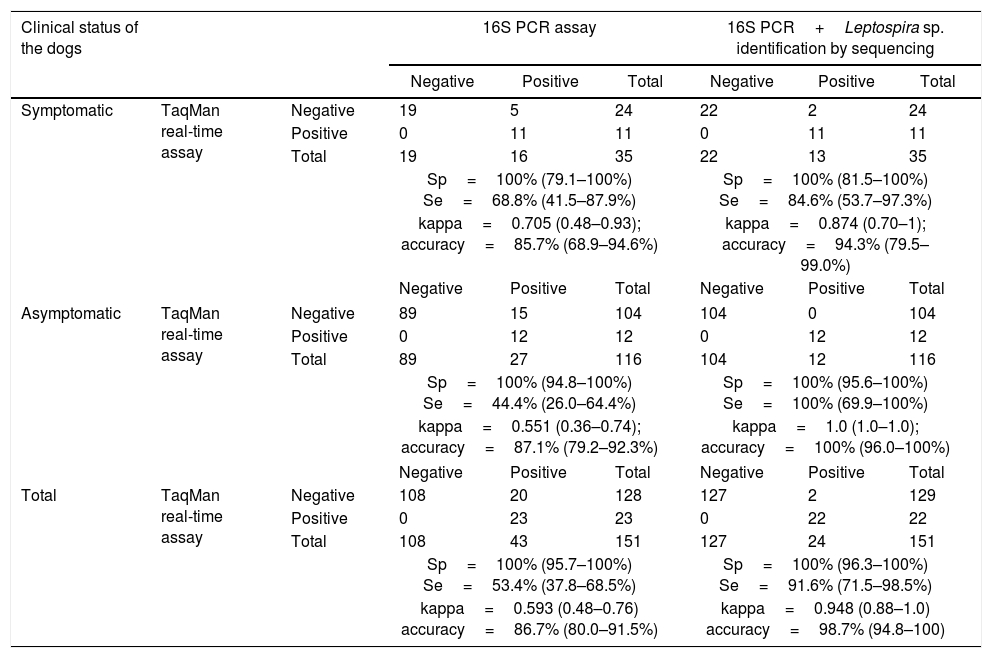
When considering both symptomatic and asymptomatic dogs, the lipl32 assay presented high overall sensitivity (91.6%; CI 71.5–98.5%), specificity (100%; CI 96.3–100%) and accuracy (98.7%; CI 94.8–100%), also revealing high agreement with the conventional PCR assay associated with sequencing analysis (kappa=0.948; CI 0.88–1). All the parameters used to compare the lipl32 TaqMan real-time PCR assay and the 16S PCR assay (with and without DNA sequencing), according to the clinical status of the dogs are presented in Table 2.
Kappa test and accuracy calculation and relative sensitivity/specificity values of the lipl32 TaqMan real-time PCR assay using the 16S PCR assay and 16S PCR assay associated with Leptospira identification by DNA sequencing as reference tests.
| Clinical status of the dogs | 16S PCR assay | 16S PCR+Leptospira sp. identification by sequencing | ||||||
|---|---|---|---|---|---|---|---|---|
| Negative | Positive | Total | Negative | Positive | Total | |||
| Symptomatic | TaqMan real-time assay | Negative | 19 | 5 | 24 | 22 | 2 | 24 |
| Positive | 0 | 11 | 11 | 0 | 11 | 11 | ||
| Total | 19 | 16 | 35 | 22 | 13 | 35 | ||
| Sp=100% (79.1–100%) Se=68.8% (41.5–87.9%) | Sp=100% (81.5–100%) Se=84.6% (53.7–97.3%) | |||||||
| kappa=0.705 (0.48–0.93); accuracy=85.7% (68.9–94.6%) | kappa=0.874 (0.70–1); accuracy=94.3% (79.5–99.0%) | |||||||
| Negative | Positive | Total | Negative | Positive | Total | |||
| Asymptomatic | TaqMan real-time assay | Negative | 89 | 15 | 104 | 104 | 0 | 104 |
| Positive | 0 | 12 | 12 | 0 | 12 | 12 | ||
| Total | 89 | 27 | 116 | 104 | 12 | 116 | ||
| Sp=100% (94.8–100%) Se=44.4% (26.0–64.4%) | Sp=100% (95.6–100%) Se=100% (69.9–100%) | |||||||
| kappa=0.551 (0.36–0.74); accuracy=87.1% (79.2–92.3%) | kappa=1.0 (1.0–1.0); accuracy=100% (96.0–100%) | |||||||
| Negative | Positive | Total | Negative | Positive | Total | |||
| Total | TaqMan real-time assay | Negative | 108 | 20 | 128 | 127 | 2 | 129 |
| Positive | 0 | 23 | 23 | 0 | 22 | 22 | ||
| Total | 108 | 43 | 151 | 127 | 24 | 151 | ||
| Sp=100% (95.7–100%) Se=53.4% (37.8–68.5%) | Sp=100% (96.3–100%) Se=91.6% (71.5–98.5%) | |||||||
| kappa=0.593 (0.48–0.76) accuracy=86.7% (80.0–91.5%) | kappa=0.948 (0.88–1.0) accuracy=98.7% (94.8–100) | |||||||
Sp: relative specificity; Se: relative sensitivity.
Out of the 35 urine samples collected from dogs presenting suspected acute leptospirosis, 11 (31.42%) showed positive results in the lipl32 TaqMan real-time PCR assay. All lipl32-positive samples also tested positive in the 16S rRNA conventional PCR; DNA from all samples could be sequenced and BLAST analysis revealed high similarity (>99%) to pathogenic Leptospira spp. However, five additional samples tested positive solely through the 16S rRNA amplification. Three of them presented high sequence similarity to non-leptospiral organisms (two in relation to uncultured bacteria and one in relation to Canis lupus familiaris), and two PCR-positive samples presented high sequence similarity to pathogenic Leptospira spp., thus revealing sensitivity differences between the lipl32 TaqMan real-time PCR assay and the 16S assay performed with or without the association with sequencing analysis (Table 2). Among the 116 samples collected from apparently healthy dogs, 12 (10.34%) tested positive in the lipl32 TaqMan real-time PCR assay; all lipl32-positive samples also tested positive in the conventional PCR and were successfully identified as pathogenic Leptospira species through further sequencing analysis (>99% similarity). Nevertheless, an additional 15 urine samples presented positive results solely through the 16S rRNA PCR (Table 2); for three samples, no readable sequences were retrieved, and sequence analyses on the other 12 samples revealed unspecific amplifications, with high sequence similarity to Collinsella intestinalis (n=2), Corynebacterium sp. (n=2), Pasteurellaceae bacterium (n=1), uncultured organisms (n=6) and Canis lupus familiaris (n=1).
All lipl32-negative samples tested positive for the MC1R gene, thus confirming that the DNA extraction and amplification procedures were appropriate.
DiscussionThe modified qPCR assay described in this study presented high analytical sensitivity, thus enabling consistent detection of low concentrations of leptospiral DNA extracted from canine urine specimens (at least 10 copies per reaction). These results were compatible with the sensitivity levels of previous studies that used the lipl32 gene as a target for molecular detection of leptospires in clinical samples.11,14,16 The reaction presented low analytical variation within or between runs, thus confirming the high analytical reliability of the assay.
The newly designed forward primer showed consensus areas in relation to all sequences retrieved from GenBank, including sequences from the two leptospiral strains belonging to L. santarosai and L. borgpetersenii that had presented nucleotide mismatches with the forward primer originally described.11 Although unusual, L. santarosai and L. borgpetersenii infections were recently reported in dogs.23,24 Consequently, development of highly sensitive assays capable of detecting a more diverse range of pathogenic Leptospira sp. could be crucial for wider application of molecular assays. The lipl32 TaqMan real-time PCR assay was able to detect DNA from all the pathogenic Leptospira spp. tested, while none of the intermediate and saprophytic strains presented amplification. Moreover, no in silico unspecific primer hybridization was detected and no amplification was observed when DNA from non-leptospiral pathogens was tested, thus confirming the high specificity of the assay.
All the positive TaqMan real-time PCR results obtained from clinical suspected cases were confirmed through the conventional 16S PCR when combined with further sequencing analysis. Despite the high overall agreement between the assays (kappa=0.874), two samples tested positive solely through the 16S PCR. The nucleotide sequences obtained from both of these samples presented high similarity to the species L. interrogans, thus confirming leptospiral infection. The performance of the TaqMan real-time PCR assay was tested several months after running the 16S PCR, and therefore the discrepancy between these results might be explained by possible loss of DNA integrity during sample storage, as previously described.25 Moreover, conventional PCR protocols are more susceptible to contamination during laboratorial procedures than are real-time assays,26 and the differences observed might also be attributable to contamination while handling the clinical material. In relation to the lipl32 TaqMan real-time PCR assay alone, 24 of the 35 dogs (68.6%) with suspected clinical leptospirosis did not present amplification of leptospiral DNA in urine samples. These results can be attributed to several causes, including: (I) intermittent urinary shedding of leptospires via urine; (II) hyperacute manifestation of the disease, thus preceding migration of the pathogen to the renal tubules; (III) clinical manifestations attributed to other causes not related to leptospiral infection; and (IV) shedding of very low quantities of leptospires in the urine, which would limit detection of the pathogen through the assay.
The molecular evaluation on urine samples taken from asymptomatic dogs showed 100% agreement between TaqMan real-time PCR and conventional PCR in association with nucleotide sequence analysis (kappa=1). Nonetheless, 15 samples tested positive solely through the conventional PCR assay, and sequencing analysis provided evidence of unspecific amplification in most of these samples (n=12). Despite several attempts, it was not possible to recover readable sequences or reproduce amplification from the other three samples, which were consequently considered negative. 16S ribosomal RNA gene sequencing is the most usual approach for molecular characterization of spirochetes and it is considered to be a well-established PCR-based tool for distinguishing leptospiral species.27 However, previous reports have shown that there are considerable risks of amplifying DNA from certain commensal and environmental bacteria,14 which undermines its use for confirmation of leptospiral infection in clinical samples. Although the 16S PCR was shown to be remarkably unspecific in the present study, the results also showed that use of primers targeting the 16S rRNA gene can provide reliable results if associated with sequencing methods. Furthermore, all unspecific amplifications in 16S PCR tested negative in the quantitative PCR, which confirmed the high specificity of the newly developed qPCR assay.
The application of the TaqMan real-time assay using combined samples from both symptomatic and asymptomatic dogs also revealed high sensitivity, specificity and accuracy standards, with high overall agreement between the newly developed TaqMan assay and the conventional PCR associated with sequencing methods, as our results have shown.
In conclusion, the present study described the development and validation of a reliable and highly sensitive quantitative assay targeting exclusively pathogenic Leptospira species. These results may contribute to a more precise diagnostic approach for identification of symptomatic and asymptomatic dogs infected by pathogenic leptospires.
Conflicts of interestThe authors declare no conflicts of interest.
We would like to thank Marcia Regina Franzolin of the Butantan Institute for providing bacterial strains. This work was supported by the Research Support Foundation of the State of São Paulo (Fundação de Amparo à Pesquisa do Estado de São Paulo, FAPESP; no. 2012/14681-7); B.A.M. holds a FAPESP grant (no. 2012/13022-0) and a PhD fellowship (no. 164284/2014) from the National Council for Scientific and Technological Development (Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq).
The following are the supplementary data to this article: