


It is known that there is correlation between biofilm formation and antagonistic activities of Bacillus subtilis strains; but, the mechanism of this correlation is not clear. So, the effect of the plant pathogen (Fusarium culmorum) on the biofilm formation in a B. subtilis strain with high antagonistic and biofilm formation activities was studied. The expression of sinR and tasA genes involved in the biofilm formation was studied in both single culture of bacterium (B) and co-culture with F. culmorum (FB) using real-time PCR. The results revealed that the expression of the sinR gene in both B and FB conditions was continuously decreased during the biofilm formation period and, after 24h (B4 and FB4), it reached 1% and 0.3% at the planktonic phase (B1), respectively, whereas the expression of the tasA was continuously increased and was 5.27 and 30 times more than that at the planktonic phase (B1) after 24h, respectively. So, the expression reduction rate for sinR (3 times) and the expression increasing rate for tasA (6 times) were significantly higher in FB conditions than the B ones. The relative expression of sinR in FB1 (planktonic phase), FB2 (8h), FB3(12h), and FB4 (24h) times was 0.65, 0.44, 0.35, and 0.29, whereas the tasA gene expression was 2.98, 3.44, 4.37, and 5.63-fold of the one at coordinate time points in B conditions, respectively. The significant expression reduction of sinR and increase of tasA confirmed that the presence of pathogen could stimulate biofilm formation in the antagonistic bacterium.
Bacillus subtilis is known as one of the most important antagonistic (biocontrol agent) and plant-growth promoting bacteria (PGPR) that is isolated from rhizosphere of different kinds of plants.1–4B. subtilis strains have the potential to produce more than two dozens of different antimicrobial compounds and antibiotics with an amazing variety of structures5 and also are able to form multicellular structures or biofilm.6–8 Biofilm formation occurs in many bacterial species in response to diverse environmental conditions such as nutrient depletion and drought, and it is mediated by many mechanical, biochemical, and genetic factors.8–10 Commonly, a mixture of polymeric compounds (e.g. extracellular polysaccharides, proteins, and DNA) and an aggregation of different microorganisms can be found in biofilms.11,12 Ability to form biofilm is associated with numerous benefits for its bacteria. For instance, antibiotics are the most common tools to remove bacteria; but, they are not efficient in the biofilm structure.6,13,14
Biofilm formation depends on two matrix gene operons, including yqxM (tapA-sipW-tasA genes) and epsA-O (15 genes) which are directly controlled by a repressor SinR and are responsible for the synthesis of amyloid-like fibers and an exopolysaccharide as two major biofilm components.2,15–19 Derepression is triggered by sinI which is activated by phosphorylated Spo0A (Spo0A∼P) as a master and important regulatory protein in biofilm formation process.2 The tasA gene in the operon yqxM is the major gene which encodes the protein involved in antimicrobial activities, spore coat assembly, and germination. It is also found in the stationary phase, sporulating cultures, and the biofilm matrix.16,17,20–22
The environmental conditions and presence of other organisms like plant pathogens, symbionts, commensalism organisms, and plant hosts can affect biofilm formation; therefore, different biofilm structures such as plaques, slimes, pellicles, and colonies are seen under various conditions.1,18,21,23,24 Previously, some researchers have shown that there is positive correlation between biofilm formation as well as PGPR and antagonistic activities of B. subtilis strains.1,2,25,26 Bais et al.1 demonstrated that a B. subtilis strain (ATCC 6051) was able to form biofilm-like structures on the roots of Arabidopsis plants and protect Arabidopsis from infections by Pseudomonas syringae. Chen et al.2 showed that plant protection by antagonistic B. subtilis strains against Ralstonia solanacearum depended on widely conserved genes required for biofilm formation, including regulatory genes and genes for matrix production; so, they provided evidence suggesting that matrix production is critical for bacterial colonization on plant root surfaces.
Previously, we isolated and selected some native B. subtilis strains which had high biofilm formation potential and antagonistic capability against Fusarium culmorum, the causal agent of foot and root rot on wheat. Finally, the strains with high biofilm production and biocontrol potential were selected. The B. subtilis strain Bs12 isolated from sugar beet fields in Kermanshah region (Iran) showed high biofilm formation, volatile production, protease activity, and 79.4% and 83% inhibitory effect against F. culmorum at laboratory and greenhouse levels, respectively.27 It was shown that volatile and protease production as well as biofilm formation by this strain and also other selected strains had significantly positive correlation with their antagonistic ability,27 which coordinated with the previous reports.1,2 The principal purpose of this investigation was to find a part of the mechanism for correlation between biofilm formation and antagonistic effect at molecular level; therefore, the effects of a plant pathogenic fungus (F. culmorum) on forming biofilm in B. subtilis (Bs12) were evaluated. To do so, expression of the tasA and sinR genes in the strain Bs12 was investigated using real-time PCR method both in the presence and absence of F. culmorum.
Materials and methodsMicroorganisms and culture conditionsThe native B. subtilis strain Bs12 (GenBank accession number HQ234328) with high potential in biofilm formation and antagonistic activity against F. culmorum was used.27 The bacterial strain was routinely grown on nutrient agar (NA) or Luria-Bertani broth (LB) at 37°C. For long maintenance, sterile 40% glycerol was used according to Weller and Cooks28 and, then, transferred to −20°C. The F. culmorum strain was kindly provided by Plant Protection Research Institute of Iran (PPRI), cultivated on potato dextrose agar (PDA) at 27°C for routine experiments, and transferred to 4°C for long time maintenance.
Primers designingTwo specific primers pairs, TasA-F (CAA GCC GTT CCA CTG TGT AG)/TasA-R (AAC CGC TCC TGA ATA TGA TGG) and SinR-F (AAA GGC TAC TCA CTA TCA GAA C)/SinR-R (TCT AAT TGA CCA TCG TAT TCG G), were designed using Oligo (National Bioscience Inc., version 5) software for conducting the real-time PCR experiments. These primers amplified 181bp and 188bp DNA fragments of tasA and sinR of B. subtilis, respectively. The primer pairs, 16SrRNA-F (GTA ACC TGC CTG TAA GAC TGG)/16SrRNA-R (CTG TAA GTG GTA GCC GAA GC), with the PCR product length of 110bp were used as the internal control. Primers were designed in order to have the length of about 20–22 bases, G/C content between 40.9% and 55%, and Tm of about 56–59°C. Length of the PCR secondary structures and dimer formation was controlled using Oligo Analyzer 1.0.3 software. The primers were synthesized by MWG (Ebersberg, Germany).
To evaluate the specificity of the primers, a PCR was performed using genomic DNA of B. subtilis (Bs12) and F. culmorum. Genomic DNA of the bacterium and fungus was extracted using GenElute™ Bacterial Genomic DNA Kit (Sigma–Aldrich, Zwijndrecht, NL) and Core-one™ kit (CoreBio, USA), respectively.
Co-culture of B. subtilis and F. culmorumBs12 cells were grown in biofilm growth medium (BGM) containing an LB-based medium plus 0.15M ammonium sulphate, 100mM potassium phosphate, pH 7, 34mM sodium citrate, 1mM MgSO4, and 0.1% glucose, as described by Hamon and Lazazzera.29 Sampling was performed under bacterial planktonic and biofilm formation conditions, according to Stanley et al.11 To obtain planktonic cells, the bacterial cells were grown overnight in BGM medium at 37°C with shaking at 200rpm. Afterwards, the medium containing bacteria was divided into two parts, one co-cultured with suspension containing 106 spores per mL of F. culmorum and another without any fungal treatment (as negative control). Both beakers were put in the above growth conditions for three more hours (OD600=2.5 for control). At this time, the bacterial medium was diluted with an OD600=0.1 in fresh medium and, then, the medium containing bacterium and fungus was diluted with the same amount of fresh medium. An aliquot of contents of each beaker as planktonic cell population (6mL) was harvested by centrifugation at 8000rpm for 10min for RNA isolation. In the second step, both beakers were incubated at 37°C without shaking; these conditions were necessary to induce biofilm formation in bacteria. The next samples containing 6mL taken from each beaker were harvested 8, 12, and 24h after incubation by centrifugation at 8000rpm for 10min. All the taken samples were immediately put at −80°C until RNA extraction. To normalize the experiments, 100μL of the media containing microorganisms were cultured on NA and bacterial CFU was counted in each sample after 24h at 37°C. Before RNA isolation, normalization was performed by diluting the samples containing more bacterial cells as the final bacterial CFU was the same for all the treatments.
Total RNA isolation and cDNA synthesisTotal RNA was extracted from the harvested samples using Aurum Total RNA Mini Kit (Bio-Rad, USA) according to the manufacturer's instructions. The concentration of RNA was quantified using a spectrophotometer (NanoDrop 1000 spectrophotometer-Thermo Scientific). PCR was performed using RNA (0.6μg) as the template to ensure the absence of genomic DNA contamination in the RNA samples. The temperature profile for PCR consisted of a first denaturation step of 5min at 94°C, followed by 40 cycles of 94°C/1min for denaturation, 60°C/1min for annealing, and 72°C/1min for extension. A final extension was carried out at 72°C/5min.
Total RNA was transformed into cDNA using iScript™ cDNA Synthesis Kit (Bio-Rad, USA) following the manufacturer's protocol. The temperature program for cDNA synthesis was 25°C/5min for the attachment of primers, 42°C/45min for cDNA synthesis, and 85°C/5min for enzyme inactivating.
Real-time PCRReal-time PCR was carried out using iCycleriQ real time PCR (Bio-Rad, USA) using IQ™ SYBR® Green Supermix Kit (Bio-Rad, USA) in 96-well plates. After the dilution of cDNA, 1μL (20ng) was added to 24μL of PCR mixture (12.5μL of IQ™ SYBR® Green Supermix, 1μL of each primer at 10pmol/μL and 9.5μL of RNase-free water). Specific cDNAs were amplified by real-time PCR using the specific primers. The real-time PCR cycling conditions were designated as follows: initial denaturation at 95°C for 2min, followed by 40 cycles of 95°C for 20s, 60°C for 30s, and 72°C for 20s, and the final extension was carried out at72°C for 5min. Fluorescence measurements were recorded during each annealing step. To establish a melting curve and confirm the primers’ specificity, an additional step starting from 50 to 95°C was performed. This step included ninety 10s cycles, in each of which there was temperature increase by 0.5°C and, at the end of each 10s, the emitted fluorescence was recorded. The efficiencies of amplifications were determined by running a standard curve by the serial dilutions of cDNA. Efficiency can be calculated by the formula: E=[10(1/−s)−1]×100, where s is the slope of standard curve. For each measurement, a threshold cycle value (CT) was determined. CT is defined as the number of cycles required for the fluorescent signal to pass the threshold (i.e. exceeds the background level). Finally, the expression of genes was calculated by formula 2−ΔΔCt.30 The results were normalized using B. subtilis16SrRNA gene as the internal gene. The ultrapure water was used instead of cDNA as a negative control and the gene expression levels were compared with the negative control.
Statistical analysisMeasures were taken for each condition by cDNA synthesized from RNA extracted from three independent cultures and performed in triplicate for each gene. Real-time PCR data analysis was performed using Bio-Rad software based on the threshold cycle (CT). Analysis of variance, comparison of means, and score of treatment groups were obtained using SAS (version 9.1) and Duncan Multiple test (p<0.01).
ResultsPrimer specificity and real-time PCR optimizationTo evaluate specificity of the designed primers, PCR was carried out using genomic DNA of B. subtilis (Bs12) and F. culmorum. When bacterial genomic DNA was used as the template, TasA-F/TasA-R and SinR-F/SinR-R primers amplified 181 and 188bp fragments, respectively. In addition, the PCR product of internal control primer was a 110bp fragment. No PCR product was observed when the fungal genomic DNA or negative control was used. After sampling and RNA extraction, PCR was performed using the samples of RNA and 16SrRNA-F/16SrRNA-R. No fragment was amplified in the samples. These results confirmed that there was no DNA contamination in the RNA samples. To determine the amplification efficiency, different serial dilutions of cDNA were used for each primer. For instance, five dilutions of cDNA from 1 to 0.0001 were used for 16SrRNA primers and, finally, cycle threshold, Tm, and standard carvers were obtained. According to this experiment, the efficiency of 16SrRNA, TasA, and SinR primers was determined as 92.75%, 99.98%, and 96.78%, respectively.
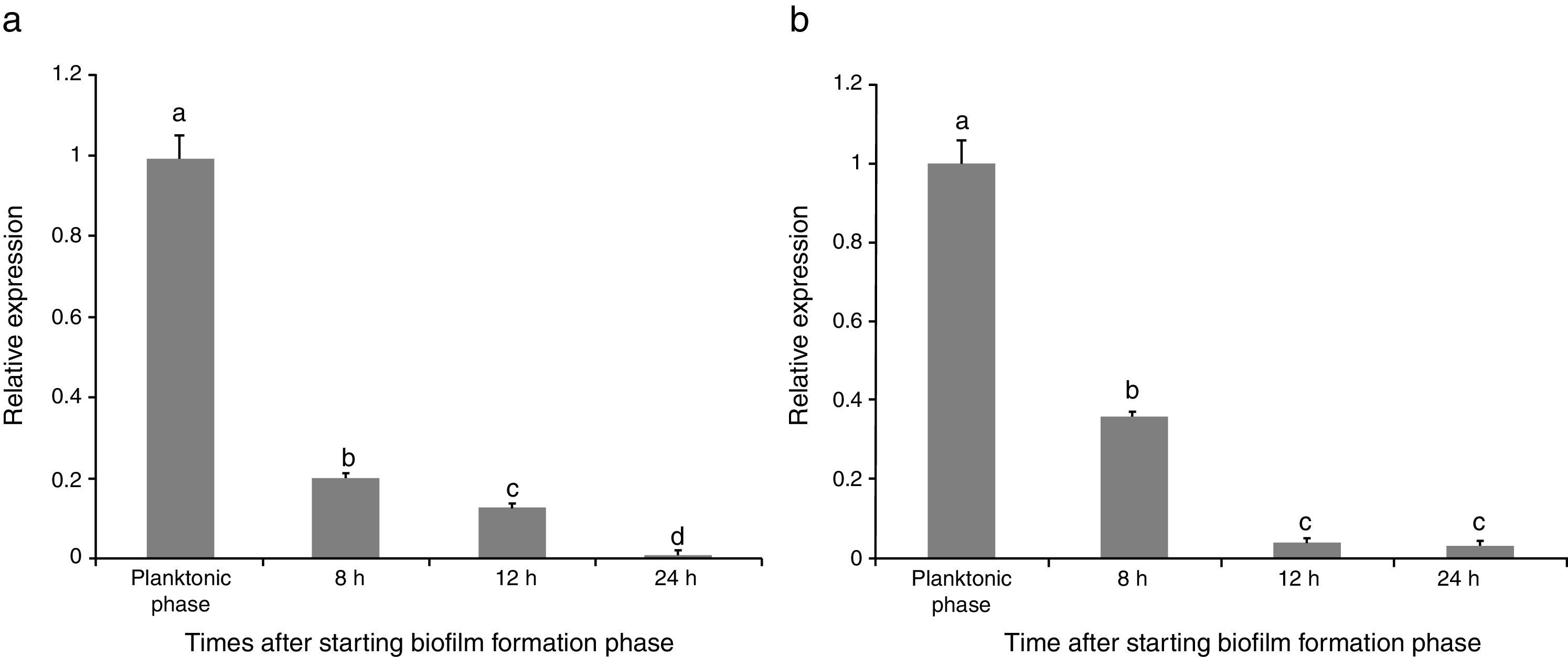
Evaluating effect of pathogen presence on sinR expressionRelative expression levels of sinR gene in the strain were calculated in the absence (B) and presence of the fungus (FB) from three independent cultures in triplicate. The results indicated that the maximum expression of sinR was observed when the bacterial cells were in the planktonic phase in the absence of F. culmorum (B1) (Fig. 1(a)). By entering the biofilm formation phase, the expression of the gene was critically decreased, which was continued over the time from B1 to B4 (24h after entering the biofilm formation period). The maximum reduction rate of the gene expression was observed 8h after starting the biofilm formation compared with planktonic phase (about 80% reductions) and the minimum expression was observed 24h after starting biofilm formation (B4) which was about 1% of the expression level in planktonic phase (Fig. 1(a)). When the bacterial cells were co-cultured with F. culmorum, the sinR expression reduction trend was critically increased. Similar to the experiments in which plant pathogen was absent, the maximum expression in the co-culture system occurred when the bacterial cells were in planktonic phase (FB1). It was continuously reduced 8 (FB2), 12 (FB3), and 24 (FB4) h after entering the biofilm formation phase and reached 3% of the expression level in planktonic phase (FB1) (Fig. 1(b)).
 in the single culture of B. subtilis strain (B), (b) in the co-culture of bacteria B. subtilis and F. culmorum (FB). Different letters indicate significant difference (p<0.05).")
Relative expression of the sinR gene during biofilm formation period compared with the planktonic phase (a) in the single culture of B. subtilis strain (B), (b) in the co-culture of bacteria B. subtilis and F. culmorum (FB). Different letters indicate significant difference (p<0.05).
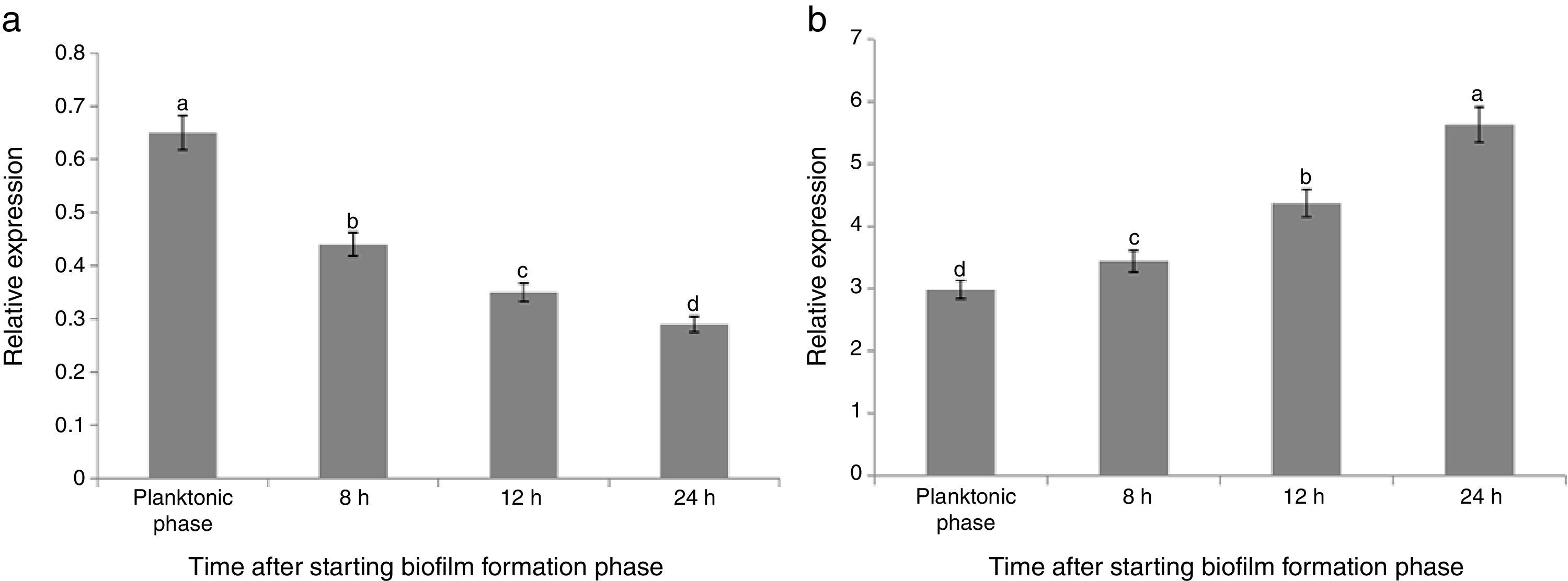
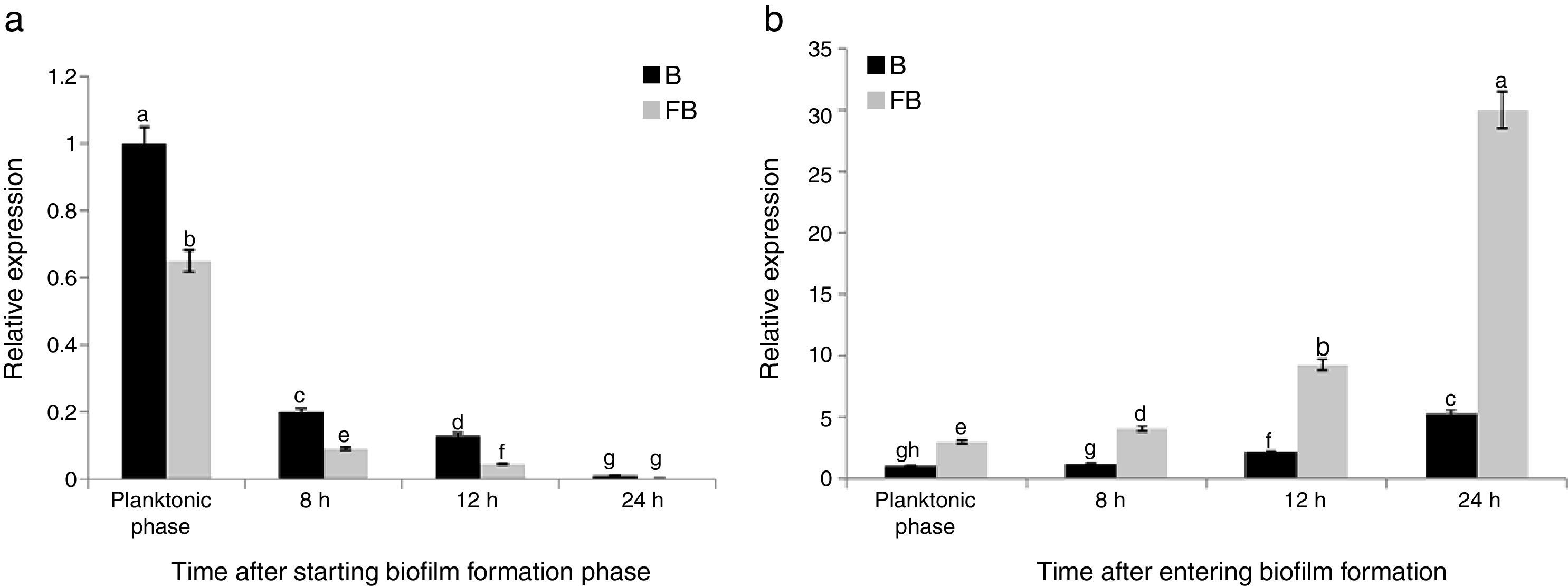
The maximum reduction rate of the gene expression was observed 8h after starting the biofilm formation (about 64% reductions). Comparison of the results of both treatments (B and FB) showed that the relative expression of sinR in FB condition was significantly lower than that of B condition in the same growth conditions and time points (Fig. 2(a)). The relative expression for FB1, FB2, FB3, and FB4 was 0.65, 0.44, 0.35, and 0.29 compared with B1, B2, B3, and B4, respectively, and by increasing the time during the biofilm formation period, the reduction rate of the sinR gene expression in FB condition was continuously increased compared with that of B condition in the same growth conditions and time points (Fig. 2(a)). The relative expression of the sinR gene during the planktonic and biofilm formation period in B and FB conditions compared with the planktonic phase of B condition is shown in Fig. 3(a). The maximum and minimum expressions were observed in the planktonic phase of B condition (100%) and 24h after starting the biofilm formation in FB treatment (0.3%), respectively (Fig. 3(a)).
 and tasA (b) genes during planktonic phase and biofilm formation periods in the co-culture of B. subtilis and F. culmorum (FB) compared with its expression point when the pathogen is absent at each time (comparison of the gene expression of FBn to Bn). Different letters indicate significant difference (p<0.05).")
Relative expression of the sinR (a) and tasA (b) genes during planktonic phase and biofilm formation periods in the co-culture of B. subtilis and F. culmorum (FB) compared with its expression point when the pathogen is absent at each time (comparison of the gene expression of FBn to Bn). Different letters indicate significant difference (p<0.05).
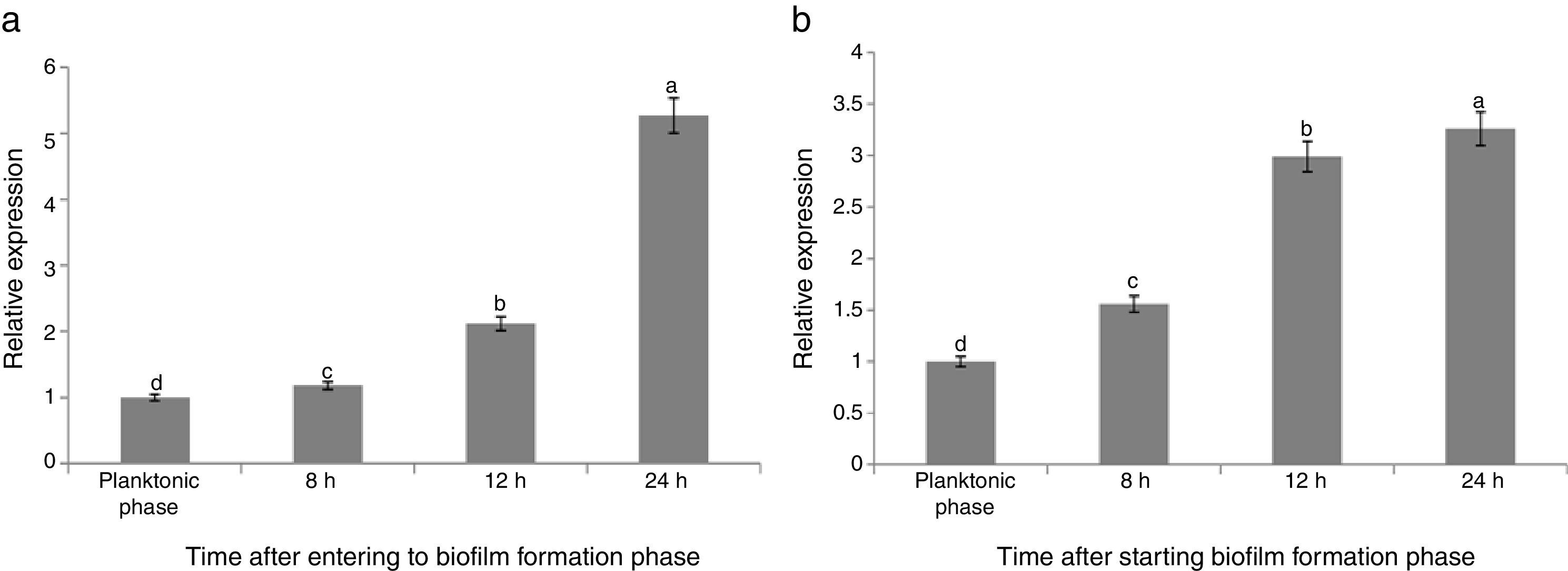
 and tasA (b) genes during biofilm formation period in both single (B) and co-culture (FB) conditions compared with the planktonic phase in the single culture condition. Different letters indicate significant difference (p<0.05).")
The results of quantitative PCR showed that the expression of tasA gene continuously increased from planktonic to the final biofilm formation phases in B condition (B1–B4). The maximum gene expression was observed when the bacterial cells were at 24h after starting the biofilm formation period in the absence of F. culmorum (B4), which was 5.27 times more than that in the planktonic phase (B1) (Fig. 4(a)). By entering the biofilm formation phase, the gene expression was critically increased, which was continued over the time from B1 to B4 phases. The maximum increasing rate of the gene expression was observed at 24h after starting the biofilm formation in which the expression of the gene was increased up to 315% compared with the phase B3, whereas the minimum increasing rate was observed at 8h after starting biofilm formation (B2) in which the expression of the gene was increased by about 18% compared with the previous phase (B1) (Fig. 4(a)).
 in the single culture of B. subtilis strain (B), and (b) in the co-culture of bacterium B. subtilis and F. culmorum (FB). Different letters indicate significant difference (p<0.05).")
Relative expression of the tasA gene during biofilm formation period compared with the planktonic phase (a) in the single culture of B. subtilis strain (B), and (b) in the co-culture of bacterium B. subtilis and F. culmorum (FB). Different letters indicate significant difference (p<0.05).
When the bacterial cells were co-cultured with F. culmorum, the tasA expression was significantly and continuously increased, which was critically increased during different phases from FB1 to FB4 (Fig. 4(b)). This increasing rate was significantly more than that of the treatments containing only bacterial cells. The levels of expression of the tasA gene 8, 12, and 24h after entering biofilm formation phase were 1.59, 2.99, and 3.26 times more than those of the planktonic phase (FB1) (Fig. 4(b)).
The expression of the tasA gene in FB1 to FB4 phases was 2.98, 3.44, 4.37, and 5.63-fold of the one at similar time points in B (B1 to B4) conditions, respectively (Fig. 2(b)). These results suggested that the pathogenic fungus stimulated the expression of tasA gene. Fig. 3(b) shows the relative expression of the tasA gene during the planktonic and biofilm formation periods in treatments B and FB compared with the planktonic phase of treatment B (B1). The maximum expression was observed 24h after starting the biofilm formation in treatment FB, which was 30 times more than that of the planktonic phase of treatment B (3000% increase) (Fig. 3(b)).
DiscussionWe characterized 30 Iranian native B. subtilis strains isolated from the rhizosphere of various hosts in different regions of Iran. The results of laboratory and greenhouse experiments showed that volatile and protease production as well as biofilm formation by some strains had significantly positive correlation with their antagonistic ability and, finally, the most powerful antagonist strains with high biofilm production were selected. Strain Bs12 isolated from sugar beet fields in Kermanshah region showed 79.4% and 83% inhibitory effect against F. culmorum at laboratory and greenhouse levels, respectively, and high biofilm formation, volatile production, and protease activity; therefore, it was selected for the present study.27 Previously, it has been shown that many different factors such as different fungal compounds (fungal culture supernatant), pH, temperature, nutrient compounds,31 indole,13 complex polysaccharides,32 and oxygen rate affect the biofilm formation. So, to explore the detailed mechanisms of different factors on biofilm formation in B. subtilis, it is necessary to perform detailed studies covering all biotic and abiotic environmental factors. Different genetic pathways that are induced by environmental signals are involved in the interaction of cells and abiotic surfaces. These factors can be changed in the amount or type of nutrient content, osmotic factor, pH, temperature, iron, oxidative stress, and substrate type.33,34 Stanley and Lazazzera35 showed that environmental signals and regulatory proteins affect the initial steps of bacterial biofilm formation and the nature of mature biofilm structure. So, surface attachment and biofilm formation on different biotic and abiotic substrates are influenced by nature and various environmental stimulations.24 The presence of other organisms, such as pathogens, is known as one of the factors affecting the biofilm formation and structure in B. subtilis; but, the mechanism is not well known.1,25,26 So, the principal purpose of this investigation was to find a part of the mechanism for correlation between biofilm formation and antagonistic effect at molecular level; therefore, the effects of a plant pathogenic fungus (F. culmorum) on forming biofilm in B. subtilis (Bs12) were evaluated. To do so, real-time PCR as a sensitive and quantitative technique was used to measure the expression profiles of two important genes (sinR and tasA) involved in the biofilm formation process of the bacteria in the presence and absence of F. culmorum, the causal agent of wheat common root rot. B. subtilis is commonly isolated from rhizosphere of different plants, shows antagonistic activities against plant pathogens, and may be used as plant-growth promoting bacteria.1,3,4 Various microorganisms can be found together in the rhizosphere. The presence and production of metabolites by other microorganisms can be very effective for biofilm formation in target bacteria. Our results indicated that the expression of sinR was significantly reduced in the presence of the pathogenic fungus. Expression of this gene was at a high level in the planktonic phase of bacterial growth; but, it decreased upon entering the production of biofilm, as was expected. Several previous studies have demonstrated that sinR as one of the most important regulatory genes has a direct negative control on biofilm formation of B. subtilis.17,20,21 Leiman et al.36 showed that point mutations in the sinR gene resulted in a significant increase in biofilm formation in B. subtilis and confirmed that it is a key matrix regulatory gene for biofilm formation. Previously, this subject has been also confirmed by other researchers.37,38 Synthesis of main components of biofilm matrix, such as extracellular polysaccharides and proteins, is mediated by two operons of 15-gene eps and three-gene yqxM, respectively.16 Both of these operons are under direct negative control of the sinR gene. Indeed, this repressor protein binds to multiple sites within the promoter region for the mentioned operons, thereby repressing its transcription. When this negative regulator is active, expression of these 18 important genes will be suppressed. So, sinR gene is known as a master negative regulator in the biofilm formation process of B. subtilis.17 Transcription of the sinR gene is controlled by another gene called sinI. When bacteria are in biofilm formation conditions, such as environmental stress, shortage of some nutrient sources, etc., transcriptional factor SpoA becomes phosphorylated and activates expression of sinI. Activated sinI can be prevented from sinR expression. Thus, eps and yqxM operons are activated and, consequently, the genes involved in biofilm formation are expressed.20 In the present study, it was observed that the expression of sinR was decreased in the biofilm compared with the phase of planktonic cells in both treatments. On the other hand, expression of sinR in free-swimming cells was higher than that of the sessile ones in the presence or absence of F. culmorum. The expression of sinR in FB condition was less at each time point compared with B condition. In planktonic cells, the gene expression level was decreased in the co-culture system (FB1) compared with single culture of bacterium (B1). This diminution rate was repeated at each time point of biofilm formation phase, namely FB2, FB3, and FB4 samples compared with B2, B3, and B4 samples, respectively. These results suggested that the presence of fungus in Bs12 growth medium caused a significant reduction of sinR gene expression in both planktonic and sessile cells.
In the case of tasA gene, opposite results were obtained. In the planktonic phase, expression of the gene was at the lowest level. By entering the biofilm production phase, the expression of the tasA gene was increased and, at final time point of biofilm formation (B4), it reached the highest levels of 5.28-fold. Previously, some other researchers have reported these results and shown significant increase in the expression of tasA gene during biofilm formation.20,39,40 This increasing rate was observed in bacterial growth phases in both single and co-culture conditions; but, the increasing rate in FB was significantly more than that of treatments B. Branda et al.16 showed that TasA is a major protein in biofilm extracellular matrix and the absence of this protein results in a residual matrix. TasA has been detected in stationary phase and sporulating cultures. It appears that TasA has several other functions; for instance, it acts as a broad-spectrum antibacterial factor and seems to have roles in spore coat assembly and germination.40,41 Evaluation of the relative expression of tasA gene showed that the expression in FB treatments was significantly increased compared with B treatments at each time point. Similar results were observed in planktonic and all biofilm formation levels. Based on these data, expression of tasA gene increased in the presence of pathogenic fungus. Many transcriptional factors in physiological activities of bacteria are regulated by environmental stress; for instance, biofilm formation by B. subtilis stimulated in non-optimal growth conditions. As F. culmorum is a pathogenic fungus, its presence in growth medium of the antagonist bacteria has provided a non-optimal condition; consequently, induced and enhanced biofilm formation down-regulate sinR and up-regulating tasA genes.
In addition to the mentioned key genes involved in biofilm formation in B. subtilis, it has been recently demonstrated that other different genes and factors affect biofilm formation. For instance, it has been shown that the genes encoding antimicrobial proteins, such as surfactin and bacillomycin which are involved in the antagonistic activities of bacterium against plant pathogens, significantly and positively affect biofilm formation.25,26 Gerwig et al.42 confirmed that the protein tyrosine kinases EpsB and PtkA differentially affected biofilm formation in B. subtilis.
Also, it has been shown that the RapP-PhrP Quorum-sensing system of B. subtilis affects biofilm formation through multiple targets due to an atypical signal-insensitive allele of RapP.43 Recently, complete genome of some biofilm-forming B. subtilis strains with antagonistic activities has been sequenced. Also, more detailed information about the correlation of antagonistic activities and biofilm formation is expected to be explored.44
In conclusion, according to our results, for the first time, it was shown that the major genes, including sinR and tasA involved in biofilm formation in B. subtilis, were significantly affected by the interaction of bacteria and fungus. Also, the presence of F. culmorum stimulated biofilm formation in B. subtilis. These findings confirmed that the presence of other organisms, such as plant pathogens in the environment of the bacterium, stimulated biofilm formation. The present study could be the first step to determine the mechanism of relationship between antagonistic activities and a biofilm formation. But, to characterize the detailed mechanisms, it is necessary to perform more detailed studies in this field.
Conflicts of interestThe authors declare no conflicts of interest.
The authors wish to thank all the staff of Department of Microbial Biotechnology and Biosafety, Agricultural Biotechnology Research Institute of Iran (ABRII), for their technical assistance. This work was supported by a grant from Agricultural Research, Education and Extension Organization (AREEO) (Grant number: 2-05-05-8711).