





Dengue is a major worldwide public health problem, especially in the tropical and subtropical regions of the world. Primary infection with a single Dengue virus serotype causes a mild, self-limiting febrile illness called dengue fever. However, a subset of patients who experience secondary infection with a different serotype can progress to a more severe form of the disease, called dengue hemorrhagic fever. The four Dengue virus serotypes (1–4) are antigenically and genetically distinct and each serotype is composed of multiple genotypes. In this study we isolated one Dengue virus 1 serotype, named BR/Alfenas/2012, from a patient with dengue hemorrhagic fever in Alfenas, South Minas Gerais, Brazil and molecular identification was performed based on the analysis of NS5 gene. Swiss mice were infected with this isolate to verify its potential to induce histopathological alterations characteristic of dengue. Liver histopathological analysis of infected animals showed the presence of inflammatory infiltrates, hepatic steatosis, as well as edema, hemorrhage and necrosis focal points. Phylogenetic and evolutionary analyses based on the envelope gene provided evidence that the isolate BR/Alfenas/2012 belongs to genotype V, lineage I and it is probably derived from isolates of Rio de Janeiro, Brazil. The isolate BR/Alfenas/2012 showed two unique amino acids substitutions (SER222THRE and PHE306SER) when compared to other Brazilian isolates from the same genotype/lineage. Molecular models were generated for the envelope protein indicating that the amino acid alteration PHE 306 SER could contribute to a different folding in this region located within the domain III. Further genetic and animal model studies using BR/Alfenas/2012 and other isolates belonging to the same lineage/genotype could help determine the relation of these genetic alterations and dengue hemorrhagic fever in a susceptible population.
Dengue virus (DENV) is an arbovirus transmitted to humans through the bite of infected Aedes aegypti mosquitoes. Dengue disease is endemic in several countries of Africa, the Americas, Mediterranean, Southeast Asia and the Western Pacific and almost half of the world's population live in risk areas for dengue.1 DENV infects 50–100 million people each year and among the infected patients 500,000 are at risk to develop the more severe diseases, such as Dengue Hemorrhagic Fever (DHF).2,3 DENV is an enveloped virus that belongs to the family Flaviviridae, genus Flavivirus. The viral genome encodes a single open reading frame which, when translated, produces a polyprotein that is processed into three structural proteins (capsid, membrane and envelope proteins) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5).
The phylogenetic analysis of DENV isolates provides insights into the evolutionary and the migratory processes of DENV, thus contributing to a better understanding of the epidemiology of the disease.4–6 In Brazil, the circulation of all four DENV serotypes has been reported and several studies have revealed a substantial genetic diversity among Brazilian DENV serotypes/genotypes. One DENV-1 outbreak was reported in Northern region of Brazil, in 1981, but it only established wider circulation in the country a few years later. DENV-1 was probably reintroduced into Brazil in 1984–19857 and in 1986 an outbreak was reported in the Southeast region. Subsequently, DENV-1 spread throughout the country. In the following years, the other 3 serotypes (DENV-2, DENV-3 and DENV-4) were also introduced into Brazil.8–10 The phylogenetic studies of Brazilian DENV-1 isolates revealed the circulation of different lineages of genotype V7,11,12 introduced into the country in different times, probably from different countries, leading to lineage replacement or to the co-circulation of different DENV-1 lineages.7
The Minas Gerais State (Southeast region of Brazil) has been gone through recurrent epidemics of dengue. DENV was first described in Minas Gerais in 1987, and since 1996, regular outbreaks have caused considerable illness in the state. During 1987–2010 the viral isolation data showed the presence of DENV-1, DENV-2, and DENV-3 in Minas Gerais and only in 2011 was DENV-4 reported in the state. Beside the circulation of all 4 serotypes in the Minas Gerais State, DENV-1 continued to be the most frequent serotype observed in 2011 and 2012.13 Despite the importance of the disease for the region, there have not been many studies that analyze the genetic variability of DENV isolates in this region. Therefore, this work aims to perform a molecular and phylogenetic analysis of a DENV-1 strain isolated in southern Minas Gerais state, Brazil.
Material and methodsStrain and sample preparationThe strain BR/Alfenas/2012 was obtained from a serum sample derived from a patient with dengue hemorragic fever, in 2012, in Alfenas, Minas Gerais State, Brazil. The patient presented skin hemorragic manifestations and laboratory findings demonstrated the increase in hematocrit concurrent with rapid decrease in platelet count. Serological tests confirmed the presence of anti-Dengue IgM and IgG (Panbio Dengue Duo IgM and IgG Capture ELISA, Alere, Australia) confirming thus, the secondary infection of this patient. Blood was collected seven days after the onset of the first symptoms upon authorization of the patient. This study was carried out by following the rules and laws that govern the use of human and animal material (approved protocol 08410912.9.0000.5142).
Cells and virus isolationAedes albopictus C6/36 cells were propagated in Leibovitz L15 medium (Cultilab, Brazil) supplemented with 10% fetal calf serum (Cultilab, Brazil) at 28°C and then used for virus isolation. For virus isolation, a 50-μL sample of serum was incubated with C6/36 cells supplemented with 2% fetal calf serum. Infected C6/36 cells showing typical cytopathic effect were harvested, and the tissue culture supernatants were used for viral RNA extraction QiAmp Viral RNA kit (QIAGEN, USA). Uninfected cells were used as negative controls.
RT-PCR and sequencing of NS5 and Envelope geneFirst reverse transcription and amplification was performed using universal primers targeting all three flavivirus subgroups as previously described.14 The region amplified encoded part of the methyltransferase and most of the RNA-dependent-RNA-polymerase domain of NS5 and, given its conserved pattern, it was chosen for flavivirus detection and identification. Second, a reverse transcription and amplification of the full envelope gene was performed using the protocol previously described.7 The envelope gene was chosen for phylogenetic and evolutionary analyses since it is a more variable gene that possesses a robust phylogenetic signal. All amplicons were sequenced using the Big Dye chemistry (USA) on ABI3730xl DNA sequencers according to Applied Biosystems protocols.
Phylogenetic and evolutionary analysesThe generated sequences were compared with sequences deposited in GenBank using the BLASTN program (www.ncbi.nlm.nih.gov/blast).15 Nucleotide sequences from envelope (total of 118 sequences, spanning 1485bp) and NS5 (total of 72 sequences of 789bp) genes were retrieved from GenBank and used for phylogenetic and evolutionary analyses. Nucleotide sequences were aligned using Clustal W program implemented in MEGA6.16 Using the software MEGA 6, the nucleotide substitution model that best fit the data was chosen (Tamura-Nei (TN93+G))17 and then it was used to estimate the evolutionary distance between sequences. Phylogenetic trees were reconstructed based on envelope or partial NS5 gene sequences, using the nucleotide substitution model TN93+G, the Maximum Likelihood method, and 1000 bootstrap replicates.18,19
Coalescent trees were also reconstructed through Bayesian analysis and the time of the most recent common ancestor (MRCA) for some strains and lineages was calculated using BEAST package v.1.8. with Markov Chain Montecarlo algorithms (MCMC).20 BEAUTI v.1.8.120 was used to create input files for BEAST, using 119 DENV-1 envelope gene sequences and the year each strain was obtained was used as the calibration point. Three different runs were performed using the nucleotide substitution model TN9317 with four gamma categories, the Bayesian Skyline method,21 under relaxed (uncorrelated lognormal) molecular clock. One hundred million chains were run (discarding the first ten million steps) and data and trees were sampled every 5000 steps. The results from three independent runs were combined using LogCombiner,22 resulting in 54,000 trees that were summarized in a maximum clade credibility tree, using TreeAnotator v 1.8.1.23 The convergence of parameters was verified with the Tracer v1.6.024 and uncertainties were addressed as the 95% highest probability density (HPD) intervals The final tree was displayed in FigTree v1.4.2.25 For phylogeographic inferences, analyses were performed as described with the addition of a geographic location attribute to each taxon and the analyses were carried out using a standard continuous-time Markov chain (CTMC) and a Bayesian stochastic search variable selection (BSSVS).26
Molecular modeling of envelope proteinMolecular models were generated for the envelope protein using MODELLER 9.12 software and also using three DENV-1 envelope proteins as templates (PDB codes: 4B03 (resolution 6 A), 4C2I (resolution 6 A) and 4GSX (resolution 1.90).27 Alignment of target and template sequences was made using the program clustalW2.28 We have generated 50 templates for each molecule, using a slow level of molecular dynamics and a variable target function method. The maximun number of interactions was equal to 500, with 2 cycles of optimization. The selected models were overlapped using the VMD program, giving prominence to the amino acid residues that differ between the proteins.29 The stereochemical quality of the model was evaluated using PROCHECK software.30
Infection of mice and histopathological analysisSix weeks old mice were infected intraperitoneally with 1.0×104 plaque forming units (pfu) of DENV-1 (n=5) and uninfected mice (n=5) was used as negative control. After 7 days of infection the animals were euthanised by cervical dislocation, and the liver removed for histopathological studies. The route of infection and time of histopathological analysis were standardized in previous studies (unpublished data). The livers were fixed with 10% formaldehyde in PBS at room temperature, dehydrated in ethanol and embedded in paraffin. Serial sections (5μm) were stained with hematoxylin and eosin. The tissue slides were examined under a Carl Zeiss Axio Scope A1 microscope attached to a Canon G10 Power Shot digital microscope camera and the captured images were analyzed with Remote Capture DC Software version 4.8. Sections were analyzed to identify the histopathological changes described in DENV infected mice.31–33
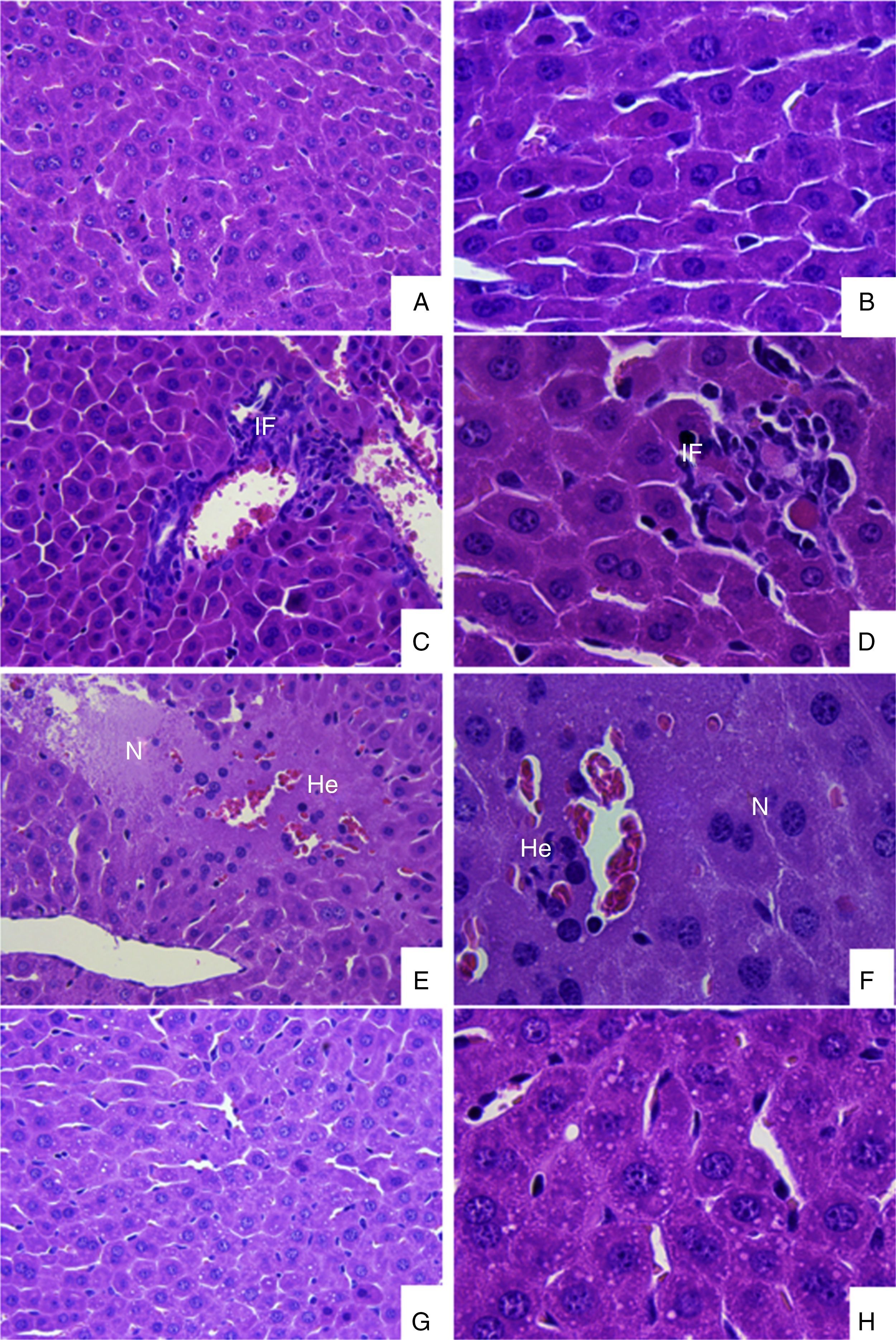
ResultsDENV-1 was isolated from a serum specimen derived from a patient with DHF, in Alfenas, in the South of Minas Gerais State, Brazil. Microscopic examination of cells inoculated with serum from the patient showed a typical cytopathic effect (CPE) in the second passage and the CPE was more intense in the fifth passage (data not show). Swiss mice were infected with BR/Alfenas/2012 in order to identify the histopathological alterations induced by the isolate. As showed in Fig. 1, the liver of infected animals showed the presence of an inflammatory infiltrate around the central veins and also focal points of edema, hemorrhage and necrosis. We also observed the presence of steatosis when compared to uninfected animals.
 infected animals. (A) and (B) Liver sections from uninfected mice in increases of 400 and 1000×, respectively; (C)–(H) Liver sections of DENV-1 infected animals demonstrating the presence of inflammatory infiltrate (C and D); focal points of hemorrhage, edema and necrosis (E and F) and hepatic steatosis (G and H). Abbreviations: IF, inflammatory infiltrate; N, necrosis; He, hemorrhage.")
Histopathological analysis of DENV-1 (BR/Alfenas/2012) infected animals. (A) and (B) Liver sections from uninfected mice in increases of 400 and 1000×, respectively; (C)–(H) Liver sections of DENV-1 infected animals demonstrating the presence of inflammatory infiltrate (C and D); focal points of hemorrhage, edema and necrosis (E and F) and hepatic steatosis (G and H). Abbreviations: IF, inflammatory infiltrate; N, necrosis; He, hemorrhage.
Cell supernatant from C6/36 cells infected with DENV-1 BR/Alfenas/2012 (fourth passage) was collected and used for total RNA extraction, RT-PCR reactions and nucleotide sequencing. Sequences corresponding to DENV envelope (GenBank KJ651912) and partial NS5 (GenBank KJ651911) were used for phylogenetic analyses. Brazilian DENV-1 strains, including DENV-1 BR/Alfenas/2012 were grouped within genotype V based on NS5 (data not shown) and E sequences analyses (Fig. 2). Brazilian strains were subdivided into different lineages and DENV-1 BR/Alfenas/2012 was clustered with DENV-1 strains from Venezuela and Brazil, detected from 1997 up to 2012. DENV-1 BR/Alfenas/2012 was closely related to strains obtained in the states of Rio de Janeiro (Southeast Brazil) and Pernambuco (Northeast Brazil).
![Phylogenetic analysis of DENV-1 strains. Bayesian coalescent analysis of DENV-1 strains was performed using a total of 119 envelope protein coding sequence (1485bp). The phylogeny and time of the most recent common ancestor (MRCA) were estimated using the year of isolation of each DENV-1 strain as the calibration point, under the relaxed molecular clock, with the Tamura Nei Model, with discrete Gamma distribution. The maximum clade credibility tree is shown, summarizing data from three independent runs. Posterior probabilities with values ≥98 are represented by an asterisk (*) and black circles in each node or branch represent the nucleotide substitution rate. The years that the some most recent common ancestors (MRCA) were estimated to exist are shown for some nodes. Sequences are initially identified by their GenBank accession number. Brazilian strains or lineages containing Brazilian strains are shown in black. For clarity purposes some branches were collapsed as follows: [BR, CO, VE (2004–2008): FJ850093, FJ639820, FJ850100, FJ639818, GQ868562, FJ639823, FJ882579, FJ639813, FJ639808, FJ639797, GQ868570, GQ868568, GU131949, GQ868569]; [BR (1986–2002): JX669475, KF672764, FJ850073, KF672763, AY277665, JX669470, JX669471, JX669474, JX669473, JX669472, JX669468, JX669467, AF311958, AF311957, JX669469, AF311956, KF672761, KF672762, AF226685, JN122280, HQ026760, AF425614]; [BR (2000–2010): HM043709, HQ026762, HQ696614, HQ026761, JX669464, GU131863, KF672759, FJ850087, JX669461, JX669465, FJ850081, FJ850075, FJ850070, FJ850090, FJ850084, FJ850077, FJ850071]; [VE, CO (1998–2005): GU131834, GU131948, FJ639743, GQ868561, GU131837, FJ639740, GQ868560]; [VE, NI, MX (1998–2009): JQ287666, FJ024479, FJ182002, GQ868530, FJ024485, FJ547088, EU596501, FJ898437, FJ898433, GQ868499, FJ810419, GU131984, GQ868500, HQ166037, FJ024483, GU131957, GU056032]; [US_PR, ARG, PAR (1993–2000): AF514878, AY277666, AY277664, FJ390380, FJ410184, EU482567] and [G1, G2, G3, G4: FJ196845, FJ196842, DQ285561, JF960217, EU863650, DQ672563, EU848545, EU081262]. BR: Brazil, VE: Venezuela, CO: Colombia, NI: Nicaragua, MX: Mexico, US_PR: Porto Rico, ARG: Argentina, PAR: Paraguay, G1, G2, G3, G4: genotypes 1, 2, 3 and 4, respectively. Analysis was carried out using BEAST package v.1.8.1, BEAUTi v.1.8.1, Tracer v.1.6.0, TreeAnotator v.1.8.1 and FigTree v.1.4.2 and LogCombiner v1.8.1.](https://static.elsevier.es/multimedia/15178382/0000004700000001/v1_201602040959/S1517838215000179/v1_201602040959/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNerEcKXq/4gYMpNkXqSlVDY0lXCTapXrEOq5lCiG1bYbW6TrxUrBaxjX0DLJ8/HpWtELMo+M3GGfm8IDry4neTHGcjD5G6d9DsRFi5VwLi1c/BK6Su/37ppfhrSNhTFfBOTbNKBpn+SP7Byq45vpV9NPbvK87o5W+rofYLaI98uDR+etotn0gSEABh8FdGwis3Iobs2PZkDfyvk+xIN6vZWKu3ote/5I7MUuyF2VfS8QiHctA023SlO6Zj3yRofXJs= "Phylogenetic analysis of DENV-1 strains. Bayesian coalescent analysis of DENV-1 strains was performed using a total of 119 envelope protein coding sequence (1485bp). The phylogeny and time of the most recent common ancestor (MRCA) were estimated using the year of isolation of each DENV-1 strain as the calibration point, under the relaxed molecular clock, with the Tamura Nei Model, with discrete Gamma distribution. The maximum clade credibility tree is shown, summarizing data from three independent runs. Posterior probabilities with values ≥98 are represented by an asterisk (*) and black circles in each node or branch represent the nucleotide substitution rate. The years that the some most recent common ancestors (MRCA) were estimated to exist are shown for some nodes. Sequences are initially identified by their GenBank accession number. Brazilian strains or lineages containing Brazilian strains are shown in black. For clarity purposes some branches were collapsed as follows: [BR, CO, VE (2004–2008): FJ850093, FJ639820, FJ850100, FJ639818, GQ868562, FJ639823, FJ882579, FJ639813, FJ639808, FJ639797, GQ868570, GQ868568, GU131949, GQ868569]; [BR (1986–2002): JX669475, KF672764, FJ850073, KF672763, AY277665, JX669470, JX669471, JX669474, JX669473, JX669472, JX669468, JX669467, AF311958, AF311957, JX669469, AF311956, KF672761, KF672762, AF226685, JN122280, HQ026760, AF425614]; [BR (2000–2010): HM043709, HQ026762, HQ696614, HQ026761, JX669464, GU131863, KF672759, FJ850087, JX669461, JX669465, FJ850081, FJ850075, FJ850070, FJ850090, FJ850084, FJ850077, FJ850071]; [VE, CO (1998–2005): GU131834, GU131948, FJ639743, GQ868561, GU131837, FJ639740, GQ868560]; [VE, NI, MX (1998–2009): JQ287666, FJ024479, FJ182002, GQ868530, FJ024485, FJ547088, EU596501, FJ898437, FJ898433, GQ868499, FJ810419, GU131984, GQ868500, HQ166037, FJ024483, GU131957, GU056032]; [US_PR, ARG, PAR (1993–2000): AF514878, AY277666, AY277664, FJ390380, FJ410184, EU482567] and [G1, G2, G3, G4: FJ196845, FJ196842, DQ285561, JF960217, EU863650, DQ672563, EU848545, EU081262]. BR: Brazil, VE: Venezuela, CO: Colombia, NI: Nicaragua, MX: Mexico, US_PR: Porto Rico, ARG: Argentina, PAR: Paraguay, G1, G2, G3, G4: genotypes 1, 2, 3 and 4, respectively. Analysis was carried out using BEAST package v.1.8.1, BEAUTi v.1.8.1, Tracer v.1.6.0, TreeAnotator v.1.8.1 and FigTree v.1.4.2 and LogCombiner v1.8.1.")
Phylogenetic analysis of DENV-1 strains. Bayesian coalescent analysis of DENV-1 strains was performed using a total of 119 envelope protein coding sequence (1485bp). The phylogeny and time of the most recent common ancestor (MRCA) were estimated using the year of isolation of each DENV-1 strain as the calibration point, under the relaxed molecular clock, with the Tamura Nei Model, with discrete Gamma distribution. The maximum clade credibility tree is shown, summarizing data from three independent runs. Posterior probabilities with values ≥98 are represented by an asterisk (*) and black circles in each node or branch represent the nucleotide substitution rate. The years that the some most recent common ancestors (MRCA) were estimated to exist are shown for some nodes. Sequences are initially identified by their GenBank accession number. Brazilian strains or lineages containing Brazilian strains are shown in black. For clarity purposes some branches were collapsed as follows: [BR, CO, VE (2004–2008): FJ850093, FJ639820, FJ850100, FJ639818, GQ868562, FJ639823, FJ882579, FJ639813, FJ639808, FJ639797, GQ868570, GQ868568, GU131949, GQ868569]; [BR (1986–2002): JX669475, KF672764, FJ850073, KF672763, AY277665, JX669470, JX669471, JX669474, JX669473, JX669472, JX669468, JX669467, AF311958, AF311957, JX669469, AF311956, KF672761, KF672762, AF226685, JN122280, HQ026760, AF425614]; [BR (2000–2010): HM043709, HQ026762, HQ696614, HQ026761, JX669464, GU131863, KF672759, FJ850087, JX669461, JX669465, FJ850081, FJ850075, FJ850070, FJ850090, FJ850084, FJ850077, FJ850071]; [VE, CO (1998–2005): GU131834, GU131948, FJ639743, GQ868561, GU131837, FJ639740, GQ868560]; [VE, NI, MX (1998–2009): JQ287666, FJ024479, FJ182002, GQ868530, FJ024485, FJ547088, EU596501, FJ898437, FJ898433, GQ868499, FJ810419, GU131984, GQ868500, HQ166037, FJ024483, GU131957, GU056032]; [US_PR, ARG, PAR (1993–2000): AF514878, AY277666, AY277664, FJ390380, FJ410184, EU482567] and [G1, G2, G3, G4: FJ196845, FJ196842, DQ285561, JF960217, EU863650, DQ672563, EU848545, EU081262]. BR: Brazil, VE: Venezuela, CO: Colombia, NI: Nicaragua, MX: Mexico, US_PR: Porto Rico, ARG: Argentina, PAR: Paraguay, G1, G2, G3, G4: genotypes 1, 2, 3 and 4, respectively. Analysis was carried out using BEAST package v.1.8.1, BEAUTi v.1.8.1, Tracer v.1.6.0, TreeAnotator v.1.8.1 and FigTree v.1.4.2 and LogCombiner v1.8.1.
Coalescent and phylogeographic analyses estimated that DENV-1 BR/Alfenas/2012, DENV-1/1266/2011/BR/RJ and DENV-1/0122_2011/BR/RJ shared the most recent common ancestor (MRCA), dating from 2008 (3.65 years before 2012, 95% HPD=2.23–5.38) (Fig. 2), probably from Southeast Brazil (data not shown). DENV-1 BR/Alfenas/2012 together with strains from Rio de Janeiro and from Pernambuco shared the MRCA dating at 2006 (5.12 years before 2012, 95% HPD=3.58–7.11). These Brazilian strains shared the MRCA with strain DENV-1/VE/BID-V2468/2008 at 2005 (6.80 years before 2012, 95% HPD=4.93–9.40), from Venezuela (Fig. 2).
The evolutionary divergence estimate between DENV-1 BR/Alfenas/2012 and the Brazilian strains DENV-1/1266/2011/BR/RJ, DENV-1/0122_2011/BR/RJ, DENV-1/15_2010/BR/RJ/2010, DENV-1/148985/BR/PE/10, DENV-1/13501/BR-PE/10 and DENV-1/12898/BR-PE-10 ranged from 1.20±0.30 to 1.64±0.36, based on envelope gene nucleotide sequences (Table 1). When DENV-1 BR/Alfenas/2012 was compared to strains from Venezuela, VE/BID-V2468/2008, VE/BID-V2254/2005, VE/BID-V3540/1997 and VE/BID-V2261/2006, the genetic distance ranged from 1.64±0.36 to 2.50±0.47 (Table 1). Surveying the nucleotide alignment, we observed that DENV-1 BR/Alfenas/2012 presents 15 unique nucleotide substitutions when compared to the closest strains from Brazil and Venezuela (DENV-1/1266/2011/BR/RJ, DENV-1/0122_2011/BR/RJ, DENV-1/15_2010/BR/RJ/2010, DENV-1/148985/BR/PE/10, DENV-1/13501/BR-PE/10 and DENV-1/12898/BR-PE-10, VE/BID-V2468/2008, VE/BID-V2254/2005, VE/BID-V3540/1997 and VE/BID-V2261/2006).
Estimates of evolutionary divergence between DENV-1 envelope gene sequences.
| [1] | [2] | [3] | [4] | [5] | [6] | [7] | [8] | [9] | [10] | [11] | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| [1] BR/ALFENAS/2012 | 0.30 | 0.31 | 0.35 | 0.34 | 0.35 | 0.36 | 0.36 | 0.35 | 0.39 | 0.47 | |
| [2] 1266/2011/BR/RJ/2011 | 1.20 | 0.12 | 0.15 | 0.16 | 0.14 | 0.16 | 0.20 | 0.21 | 0.24 | 0.33 | |
| [3] 0122_2011/BR/RJ/2011 | 1.27 | 0.20 | 0.20 | 0.19 | 0.19 | 0.20 | 0.24 | 0.25 | 0.28 | 0.37 | |
| [4] 15_2010/BR/RJ/2010 | 1.47 | 0.34 | 0.55 | 0.15 | 0.14 | 0.15 | 0.18 | 0.19 | 0.22 | 0.31 | |
| [5] 14985/BR-PE/10/2010 | 1.64 | 0.41 | 0.62 | 0.34 | 0.12 | 0.14 | 0.19 | 0.18 | 0.22 | 0.30 | |
| [6] 13501/BR-PE/10/2010 | 1.57 | 0.34 | 0.55 | 0.27 | 0.20 | 0.12 | 0.18 | 0.17 | 0.22 | 0.30 | |
| [7] 12898/BR-PE/10/2010 | 1.64 | 0.41 | 0.62 | 0.34 | 0.27 | 0.20 | 0.20 | 0.19 | 0.23 | 0.31 | |
| [8] VE/BID-V2468/2008 | 1.64 | 0.55 | 0.76 | 0.48 | 0.55 | 0.48 | 0.55 | 0.13 | 0.22 | 0.28 | |
| [9] VE/BID-V2254/2005 | 1.65 | 0.56 | 0.77 | 0.48 | 0.55 | 0.48 | 0.55 | 0.27 | 0.15 | 0.23 | |
| [10] VE/BID-V3540/1997 | 1.88 | 0.77 | 0.99 | 0.69 | 0.76 | 0.69 | 0.76 | 0.62 | 0.34 | 0.27 | |
| [11] VE/BID-V2261/2006 | 2.50 | 1.35 | 1.57 | 1.27 | 1.34 | 1.27 | 1.34 | 1.05 | 0.76 | 0.97 |
The number of base substitutions per site between sequences are shown. Standard error estimates are shown above the diagonal in bold and italic.
DENV-1 BR/Alfenas/2012 presented a conservative aa substitution (ARG 394 LYS), in position 394 of the deduced amino acid envelope protein sequence (domain III). This conservative aa substitution was also observed for other sequences, but some strains (DENV-1/BR/1266/2011/BR/RJ/2011, DENV-1/BR/0122_2011/BR/RJ/2011, DENV-1/BR/15_2010/BR/RJ/2010, DENV-1/BR/14985/BR-PE/10/2010, DENV-1/BR/13501/BR-PE/10/2010, DENV-1/BR/12898/BR-PE/10/2010, VE/BID-V2468/2008, VE/BID-V2254/2005, VE/BID-V3540/1997 and VE/BID-V2261/2006) possessed an arginine residue. Moreover, DENV-1 BR/Alfenas/2012 possessed two unique amino acid substitutions, at positions 222 (domain II) and 306 (domain III), when compared to other Brazilian isolates. At position 222, one conservative amino acid substitution (SER 222 THR) was observed in DENV-1 BR/Alfenas/2012 sequence where threonine was observed. One non-conservative aa substitution took place at position 306 (PHE 306 SER) where DENV-1 BR/Alfenas/2012 exhibited a serine residue. In a second search in GenBank, looking for sequences that might also possess the same amino acid residue in this position, it was observed that one DENV-1 isolated from Viet Nam (D1.QuyNhon.29.06 – KC861922), obtained in 2006, also had the same amino acid residue in this position.
The molecular model of E protein from isolate BR/Alfenas/2012 in superposition with the isolate DENV-1/BR/0122_2011/BR/RJ/2011 showed a highly similar three dimensional model (data not showed). The aa substitutions SER 222 THR and ARG 394 LYS does not affect the predicted protein structure, since these are conservative substitutions between amino acids with very similar characteristics (Fig. 3). The other mutation (PHE 306 SER) would likely have a more pronounced effect in the predicted protein structure. The change of PHE by SER could decreases the interaction with several other residues (SER305, LYS307, LYS325, VAL380, VAL324, ILE335, TYR326 e GLY381) of envelope protein which can contribute to a different folding in this region (Fig. 3).
Discussion compared to the equivalent interactions of the mutated residue SER306 in the isolate BR/Alfenas/2012 (right).")
In 2012, a serum sample from a suspected dengue patient with hemorrhagic manifestations was sent to our laboratory in Unifal-MG. Laboratory tests and the clinical manifestations classified this patient as having grade II DHF. After viral isolation, the amplification, sequencing and analysis of NS5 gene (partial sequence) confirmed that BR/Alfenas/2012 was a DENV-1 isolate. Previous studies have demonstrated that genotype V is the only DENV-1 genotype in Brazil4,7,11,12 with different lineages circulating in the country.7,11,12 The same pattern was observed after our phylogenetic analyses based on the envelope gene where Brazilian DENV-1 strains were grouped into different lineages from genotype V, including BR/Alfenas/2012, which was also grouped within lineage I.7
DENV-1/BR/Alfenas/2012 presented a close relationship with strains from Brazil and Venezuela, sharing the MRCA with Venezuelan strains dating at 1994, confirming previous estimations.7 The Venezuelan strain that was closest to Brazilian strains was VE/BID-V3468/2008, which shared the MRCA with Brazilian strains at 2005, also as previously estimated.7 Within that lineage, DENV-1 BR/Alfenas/2012 was most closely related to strains from Rio de Janeiro, a neighbor state in Southeast Brazil. The results indicated that DENV-1 BR/Alfenas/2012 is derived from strains circulating in Southeast Brazil, most probably from Rio de Janeiro, based on the available information. The MRCA of DENV-1 BR/Alfenas/2012 and two strains from Rio de Janeiro dated from 2008, indicating that the former strain might have been introduced into the southern region of Minas Gerais state, a few years before this strain was isolated from a patient with DHF, in 2012.
The evolutionary distance between DENV-1 BR/Alfenas/2012 and closely related strains (as shown in Table 1 and Fig. 2) indicated that this strain had a local evolution, resulting in a more genetically divergent strain. This is supported by the higher nucleotide substitution rate of this strain when compared to other DENV-1 strains and by the 15 unique nucleotide substitutions observed in DENV-1 BR/Alfenas/2012 envelope gene sequence (supplementary data) and three amino acid substitutions observed in the deduced amino acid envelope protein sequence.
Alignment of the deduced aa sequences for the envelope protein of the isolated BR/Alfenas/2012 with the other isolates indicates the presence of two unique amino acids substitutions that were described for the first time in Brazil. Many studies have shown that mutations that affect the E protein may alter the virulence of DENV, because these mutations may modify the viral particle interaction with surface receptors on the host cell or even interfere with the DENV fusion process with the host cell membrane.34–37 The first amino acid substitution was observed in the residue 222 and this substitution was also described in DENV-1 isolated in Philippines (South Pacific), which belonged to genotype IV and fixed in all isolates of DENV-1 obtained in this country since 2002. The Residue 222 is localized in domain II, which is implicated in the dimerization of the envelope protein at acidic pH preceding the membrane fusion and viral entrance into the host cell. However, the authors did not find any relationship with clinical severity of human infection and so they suggested that SER 222 THR is not a virulence determinant.
The other amino acid substitution was observed in the residue 306 and it is located in the domain III of protein E. This domain is involved in receptor binding with the exposed residues on the surface of the virus, being responsible for determining receptor specificity, vector type and host and tropism of cells.38–40 The segment located between residues 298 and 397 have been shown to be the target of a number of neutralizing antibodies, and aa substitutions in this region could be related with a escape of neutralizing antibodies.41 The presence of this aa substitution and the differences observed in the molecular model could strengthen the role of this substitution to potentially escape neutralizing antibodies.
It is known that the introduction of new serotypes and/or genotypes/lineages of DENV is an important risk factor for dengue fever outbreaks. Susceptibility of a population to a specific serotype and the occurrence of secondary infections in a hyperendemic country have also very important role to understanding the epidemiology of the disease. Different DENV serotypes, genotypes and even lineages can be introduced into new areas increasing the genetic diversity of viral population. In addition, clonal evolution has been assumed to be most important factor in DENV evolution.6 In this scenario, monitoring the DENV is of great importance to observe the spread of potentially virulent isolates, as well as to assess their evolution and impact on the population during an outbreak/epidemic.
This is the first report of DENV-1 isolation and characterization in the southern region of Minas Gerais state, Brazil, associated with DHF. BR/Alfenas/2012 was isolated from a patient with DHF, during a secondary DENV infection and phylogenetic and molecular analyses indicated that this virus belongs to a lineage of DENV-1, genotype V, recently introduced into the country, probably from Venezuela. BR/Alfenas/2012 is most probably originated from Rio de Janeiro, but after its introduction in a new region, this strain underwent local evolution, exhibiting a higher nucleotide substitution rate, leading to this more genetically divergent strain. However, additional studies should be done to determinate the relationship of these genetic alterations in terms of having the increased potential to induce DHF in a susceptible population.
Conflicts of interestThe authors declare no conflicts of interest.
The authors thank National Counsel of Technological and Scientific Development (CNPq)/Brazil for the financial support (grant number 404005/2012-8).
The following are the supplementary data to this article: