




The selection of suitable reference genes is crucial for accurate quantification of gene expression and can add to our understanding of host–pathogen interactions. To identify suitable reference genes in Pandora neoaphidis, an obligate aphid pathogenic fungus, the expression of three traditional candidate genes including 18S rRNA(18S), 28S rRNA(28S) and elongation factor 1 alpha-like protein (EF1), were measured by quantitative polymerase chain reaction at different developmental stages (conidia, conidia with germ tubes, short hyphae and elongated hyphae), and under different nutritional conditions. We calculated the expression stability of candidate reference genes using four algorithms including geNorm, NormFinder, BestKeeper and Delta Ct. The analysis results revealed that the comprehensive ranking of candidate reference genes from the most stable to the least stable was 18S (1.189), 28S (1.414) and EF1 (3). The 18S was, therefore, the most suitable reference gene for real-time RT-PCR analysis of gene expression under all conditions. These results will support further studies on gene expression in P. neoaphidis.
Pandora neoaphidis is one of the most important fungal pathogens of aphids and has great potential for use in biocontrol.1–4 Although the life cycle of P. neoaphidis is well known, a detailed molecular understanding of gene expression during the infection process is still lacking. A diversity of genes can be up- or down regulated during host–pathogen interactions,5 however, little is known about the expression of reference genes in this particular entomophthoralean fungus.
Due to its high throughput capacity, sensitivity and specificity, quantitative real time reverse transcription polymerase chain reaction (qRT-PCR) represents a good method for the measurement of gene expression levels across different samples.6,7 Indeed, it has been used for this purpose with several fungi.8–11 However, a number of critical aspects must be optimized for effective qRT-PCR analysis; these include the efficiency of RNA extraction, the quality of the RNA, the presence of inhibitors, the efficiency of the reverse transcription and the selection of a suitable reference gene as an internal control.12 While the majority of these potential sources of error can be avoided by adhering closely to standardized protocols, selection of appropriate reference genes is frequently the greatest challenge because it requires a species-specific solution.13,14 An ideal reference gene should have constant expression across all samples to be investigated, regardless of biotype, developmental stage or any other biological or experimental variability.
Selection of an inappropriate reference gene with variable expression, leads to erroneous calculations of the expression of target genes and, therefore, incorrect assumptions about the function of those target genes.15 Identification of suitable reference genes is essential for accurate transcript expression analysis. Several statistical algorithms have been developed to identify the most suitable internal controls with the least variability in expression; these algorithms are based on qRT-PCR data from a given set of candidate genes and rank putative reference genes according to their expression stability, thereby indicating the best reference gene or combination of reference genes for accurate normalization. The four most commonly used algorithms for assessing the appropriateness of reference genes are geNorm,16 NormFinder,17 Best-Keeper18 and Delta Ct.19 These software packages are freely available to download from the authors’ websites and have been widely used to identify suitable reference genes.20,21
In this study, three putative housekeeping genes (18S, 28S and EF1) were evaluated as potentially reliable reference genes in P. neoaphidis using qPCR. Fungal infection of the host usually involves multiple developmental stages.22 Therefore, profiling gene expression in multiple development stages is important for understanding the mechanisms of pathogenesis. We tested four developmental stages including conidia, conidia with germ tubes, short hyphae and long hyphae. Profiling gene expression under various nutritional conditions is also a routine approach to study gene function. We evaluated the stability of gene expression in three different nutrient media. The expression stability of each gene in all samples was analyzed using the geNorm, NormFinder, Best-Keeper and Delta Ct programs, and the most suitable reference gene for accurate normalization was selected.
Material and methodsIsolate and culture conditionsP. neoaphidis isolate ARSEF 5403 was obtained from the USDA Collection of Entomopathogenic Fungal Cultures, Ithaca, NY. It was subcultured on SEMA (Sabouraud dextrose agar [SDA] supplemented with egg yolk and milk23; in 9cm diameter Petri dishes for 10d at 18°C in a 12:12 light:dark regimen.
Sample preparationsPropagules at different stages of germination were prepared. Mycelial mats from liquid culture were produced using the method described by Xu and Feng.24 Primary conidia actively discharged from the mycelial mat during the period of peak sporulation were harvested into 0.01mol/L sterile phosphate buffered saline (PBS) solution (130mmol/L NaCl, 7mmol/L Na2HPO4, 3mmol/L NaH2PO4, pH 7.3).25 The conidial suspension was filtered through glass wool to remove any mycelia and the conidia centrifuged at 4500rpm for 10min before being inoculated into GLEN medium in flasks26 at a concentration of 1010 conidia per milliliter followed by incubation at 20°C and 150rpm in a 12:12 light:dark regimen for different periods of time.2 Samples were taken after ∼0h for conidia, after ∼6h for conidia with germ tubes, after ∼12h for early, short hyphae (i.e. the length of germ tube was a maximum of 200μm), and after 24h for elongated hyphae (i.e. hyphae that exceeded 200μm). At each stage the fungal structures were observed microscopically to ensure they had attained the correct developmental stage (Fig. 1). Samples of each developmental stage were collected after vacuum filtration through Whatman 54 paper and stored at −80°C until total RNA was isolated. There were three replicate flasks for each developmental stage, i.e. 12 flasks in total were set up.
, conidia with germ tubes at 6h (b), early hyphae at 12h (c) and elongated hyphae at 24h (d). Propagules were stained with lactophenol and observed under a microscope. Scale bar: 50μm.")
Views of P. neoaphidis propagules. To prepare propagules at different developmental stages, primary conidia produced by a mycelial mat were incubated in GLEN broth. The cultures were centrifuged to harvest conidia at 0h (a), conidia with germ tubes at 6h (b), early hyphae at 12h (c) and elongated hyphae at 24h (d). Propagules were stained with lactophenol and observed under a microscope. Scale bar: 50μm.
To provide samples grown under different nutritional conditions, the mycelium from half a Petri dish (agar medium removed using a scalpel) was added to 100mL flasks containing either 30mL GLEN medium,26 30mL of Grace's medium (Invitrogen, USA) or 30mL of OS-SDB medium (Sabouraud Dextrose Broth [SDB; Difco, BD, USA] supplemented with 0.5% (v/v) sesame oil and 0.1% (w/v) sugar esters of fatty acids [Emulsifier E473, CAS No. 37318-31-3, Liuzhou Gaotong Food Chemicals Co. Ltd., China]). Flasks were incubated at 20°C and 150rpm in a 12:12 light:dark regimen for three days.2 Mycelium samples grown under different nutritional conditions were collected by vacuum filtration through Whatman 54 paper and stored at −80°C until total RNA was isolated. There were three replicate flasks for each nutritional condition i.e. nine flasks in total.
Candidate gene and primer designNucleotide sequences of the 18S and 28S genes (Genbank accession numbers HQ677591.1 and EF392405.1, respectively), were downloaded from Genbank (http://www.ncbi.nlm.nih.gov/genbank/), while the EF1 gene sequence was acquired through degenerate PCR. Primer pairs for each gene were designed using the Primer Premier 5.0 program (Table 1). Specificity of primer pairs for each candidate gene was confirmed using melting curve analysis and agarose gel electrophoresis.
Primer sequences of selected reference genes for qRT-PCR in P. neoaphidis.
| Gene | Accession number | Primer pairs | Amplicon size (bp) |
|---|---|---|---|
| 18S | HQ677591.1 | F:CAAACCCGAGCAATAGTCR:GTAGGAGCCCGATAGTAAA | 179 |
| 28S | EF392405.1 | F:TCTTATCAGTCTCAGCACCTTGR:TCTGTCAATCCTTACTATGTCTGG | 181 |
| EF1 | DQ275343.1 | F:GACGCACTCCTTGTTGATR:CCTTGCCACTCTGGTTCT | 82 |
RNA was prepared for each sample using the RNeasy Mini Kit (QIAGEN, Cat. No. 74104, Mississauga, Canada), and genomic DNA was eliminated by loading RNase-free DNase I into the filter column (QIAGEN). Nucleic acid extraction was performed according to the manufacturer's instructions. The quality of the RNA was checked by electrophoresis on 1% agarose gels to reveal any contaminating genomic DNA and intact rRNA subunits 28S and 18S. Nucleic acid concentrations were measured using a Nanodrop 1000 (Thermo Scientific, Wilmington, DE, USA).
First-strand cDNA was synthesized using random hexamer primers and SureStart Taq DNA polymerase according to the manufacturer's instructions (Agilent Technologies, Cat. No.20820, Santa Clara, CA, USA). To avoid errors caused by contamination with genomic DNA, all RNA samples were treated with RNase-free DNase I before reverse transcription (Agilent Technologies, USA). One microgram of total RNA was used for cDNA synthesis; the cDNA was subsequently diluted with nuclease-free water (QIAGEN) to 20ng/μL. The cDNA mixtures were diluted 1:10 with nuclease-free water (QIAGEN) and stored at −20°C for subsequent quantitative PCR analysis.
Quantitative PCRReal time RT-PCR amplification mixtures (25μL) contained 20ng template cDNA, 2× Brilliant IISYBR Green master mix buffer with fluorescein for dynamic well factor collection (12.5μL; Agilent Technologies) and 400nmol/L each of the forward and reverse primers (Eurofins Genomics, Ebersberg, Bayern, Germany). The reaction was performed using a Stratagene Mx3000P® system. PCR was accomplished after a 10min activation/denaturation step at 95°C, followed by 40 cycles of 30s each at 95°C, 60s each at 60°C and 30s each at 72°C. Fluorescence was detected at each polymerization step. To test the PCR efficiency of each primer pair, a cDNA mixture containing equal amounts of cDNA from all samples was used as the template. The ten-fold dilution series of the cDNA mixture (from 20ng to 2pg) was used as the template. After the PCR amplification, melt curve analysis identified any production of dimers. All PCR amplifications were conducted from replicate samples and in duplicate.
Statistical analysesThe samples were divided into two groups: different developmental stages (12 samples) and different nutritional conditions (9 samples). The expression levels of the candidate reference genes were determined from the cycle threshold (Ct) value, the number of PCR cycles at which the quantity of amplified targets reached a specific threshold level of detection. The expression stability of the reference genes was evaluated using the four programs, geNorm,16 NormFinder,17 BestKeeper18 and Delta Ct19 to analyze all the raw Ct values from the two groups; in order to balance the ranking of the candidate reference genes across the different algorithms, expression stability was evaluated in the program http://www.leonxie.com/referencegene.php?type=reference, to determine an overall comprehensive stability value for each candidate reference gene.
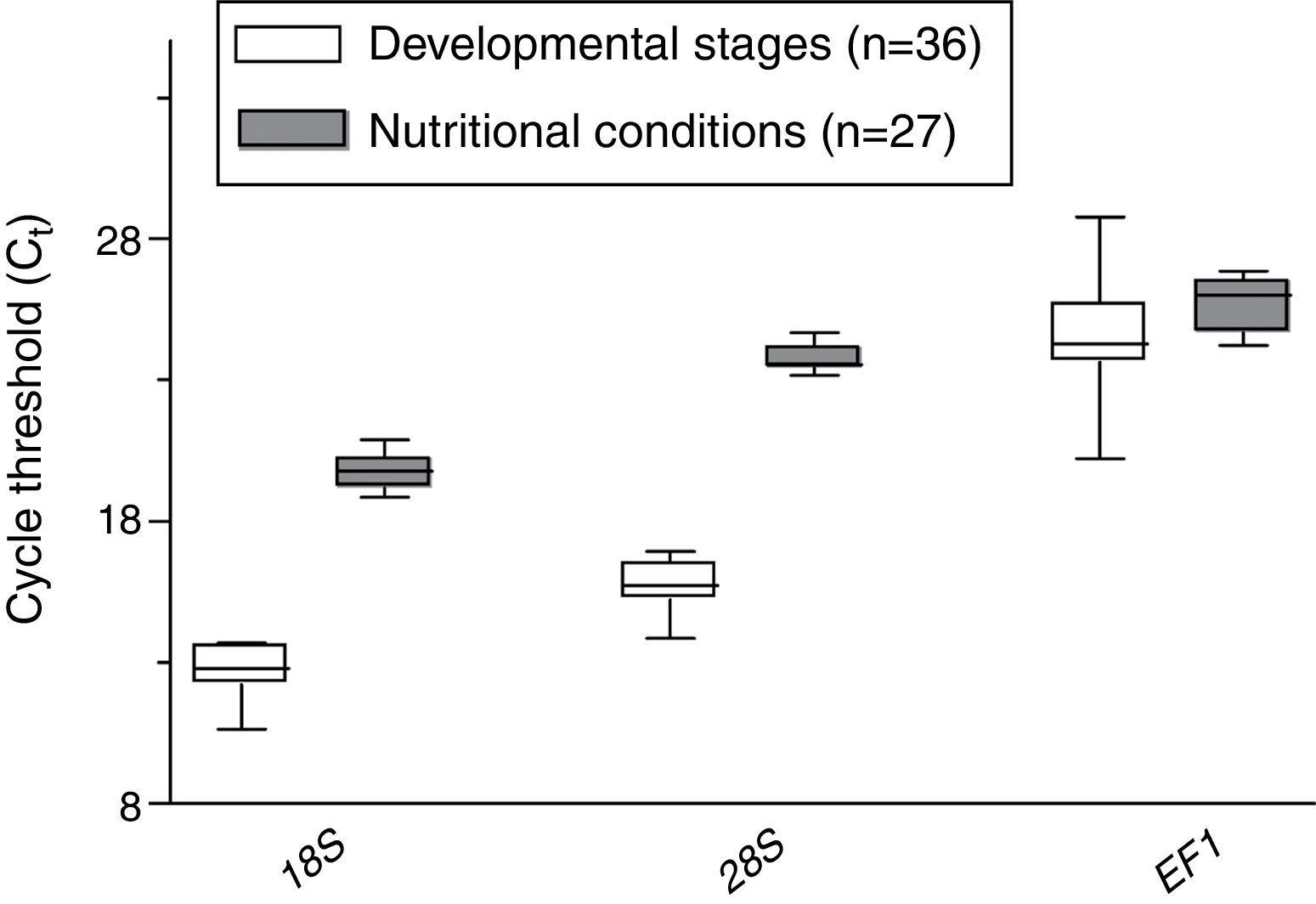
ResultsExpression profiling of candidate reference genesThe calculated expression levels (Ct values) of the three genes evaluated varied from 10.65 to 28.75, which represents quite a large degree of variation (Fig. 2). The 18S gene was identified as the most abundant transcript in all samples; the mean Ct value for the 18S gene from different developmental stages and different nutritional conditions was 12.62 and 19.76, respectively. The mean Ct value for the 28S gene from different developmental stages and different nutritional conditions was 15.67 and 23.74, respectively. In contrast, the mean Ct value for the EF1 gene from different developmental stages and different nutritional conditions was 24.25 and 25.65, respectively.
Candidate gene expression stability of different developmental stages and under different nutritional conditions
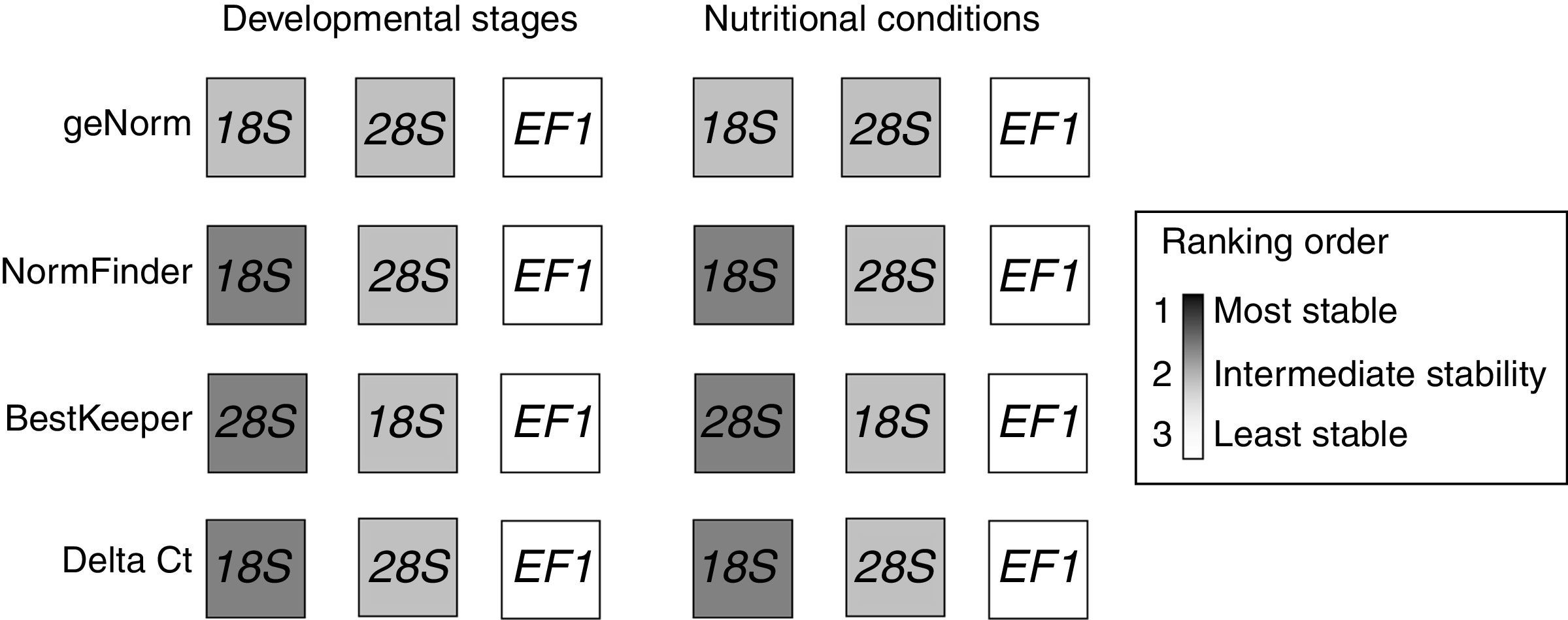
Using the algorithms geNorm, NormFinder and Delta Ct, for developmental stage and nutritional conditions, the 18S gene was the most stable of the three genes, followed in rank order by the 28S gene and finally the EF1 gene (Fig. 3). However, using the BestKeeper algorithm, the 28S gene was much more stable than the 18S or EF1 genes (Fig. 3).

Comparison of the ranking in expression stability calculated by geNorm, NormFinder, BestKeeper and Delta Ct. The analysis was performed separately for each sample group under the conditions examined and the ranking orders were highlighted by different gray tones from white to dark, darker gray indicates greater stability.
Specifically, using geNorm software analysis, the P. neoaphidis expression stability values at different developmental stages and under different nutritional conditions for the three candidate reference genes (M value) were in the order: 18S (0.457)>28S (0.534)>EF1 (0.749) and 18S (0.389)>28S (0.557)>EF1 (0.607), respectively. Using NormFinder software analysis, the P. neoaphidis expression stability values at different developmental stages and under different nutritional conditions for the three candidate reference genes (stable value) were in the order: 18S (0.084)>28S (0.264)>EF1 (0.509) and 18S (0.118)>28S (0.355)>EF1 (0.403), respectively. Using Delta Ct software analysis, the P. neoaphidis expression stability values at different developmental stages and under different nutritional conditions for the three candidate reference genes (stable value) were in the order: 18S (1.19)>28S (1.20)>EF1 (2.02) and 18S (0.41)>28S (0.55)>EF1 (0.59), respectively.
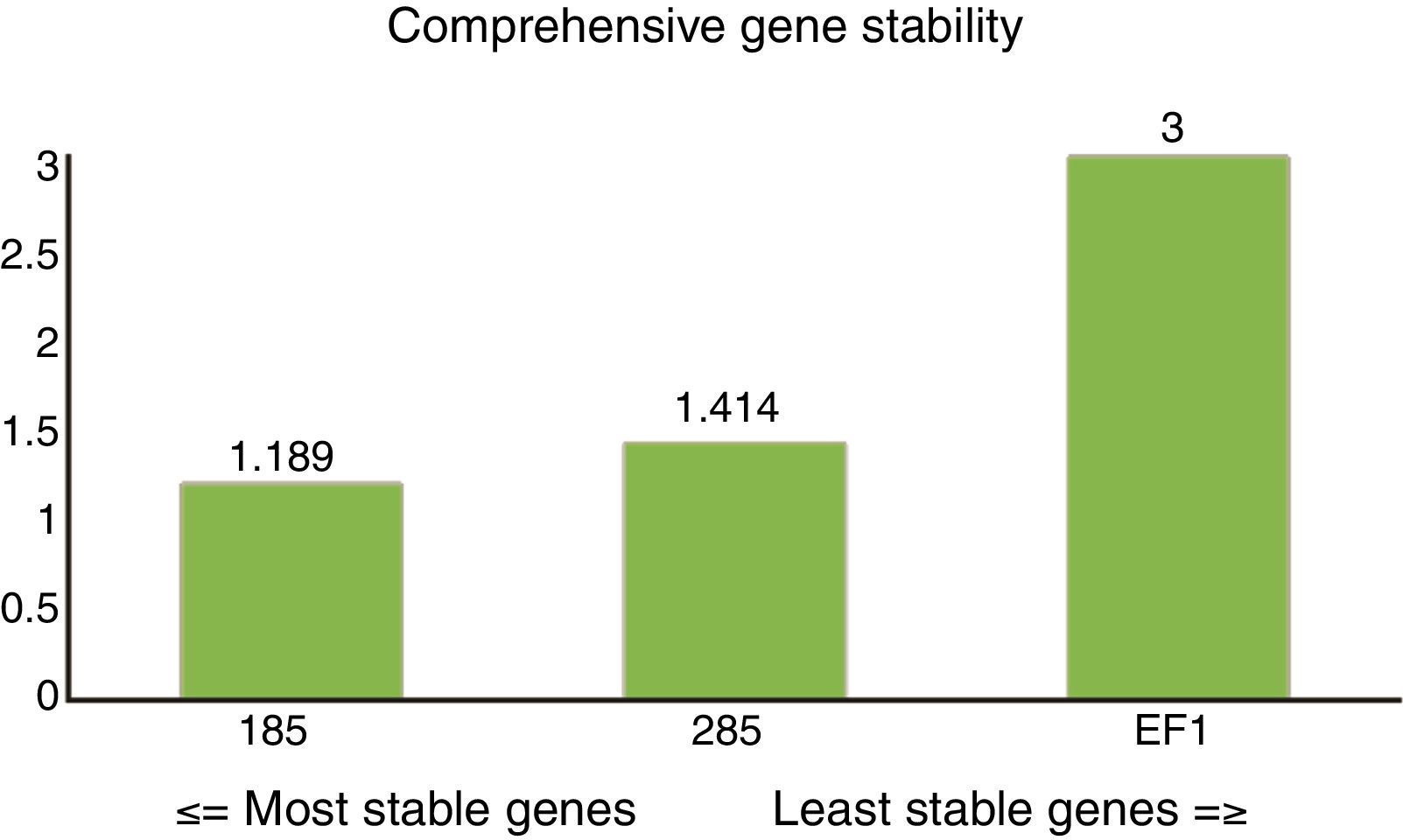
Overall stability ranking of candidate reference genesThe ranking from the comprehensive gene stability analysis of candidate reference genes was 18S (1.189)>28S (1.414)>EF1 (3.000) (Fig. 4). The 18S gene could, therefore, be selected as the most stable reference gene for normalization of qRT-PCR.
Discussion
qRT-PCR is becoming more and more important as a research tool to identify and characterize genes that are differentially expressed during different types of growth and at different developmental stages.7 An appropriate reference gene or set of reference genes are required for accurate gene expression analysis and, before this study, were not available for P. neoaphidis.
Due to a lack of sequence data for the genus Pandora, we selected three housekeeping genes (18S, 28S and EF1) as candidate reference genes for expression studies in P. neoaphidis (Table 1). We did not include the beta-tublin (β-tublin) reference gene because the primers we developed for it also amplified the host aphid's cDNA which would preclude its use in gene expression studies of the interaction between the fungus and the host aphid (unpublished data). Prior to doing the qPCR, we checked the specificity of the designed primers; analysis of the melting curves showed an absence of dimers in the PCR amplifications.
Endogenous controls commonly include the use of single, constitutively expressed, housekeeping genes. However, the expression of a single housekeeping gene can vary considerably between samples27–29 and may not, therefore, be acceptable for normalization of real-time RT-PCR. This is the reason that we wanted to select genes with reliable and accurate expression levels that were expressed consistently at different developmental stages (primary conidia, conidia with germ tubes, early hyphae and elongated hyphae) and under different nutritional conditions (GLEN, Grace's medium and OS-SDB).
If only one software package, with one algorithm, is used to analyze the biological stability of expression in candidate reference genes it is often difficult to select the overall best reference gene.30 Therefore, in this study, a comprehensive online tool based on four different algorithms (geNorm, NormFinder, BestKeeper and Delta Ct) was adopted. The results for geNorm, Normfinder and Delta Ct were all very similar and showed that, of the three candidate reference genes, expression of the 18S gene was the most stable; this result is consistent with a number of other reports.31,32 However, the stability calculated by BestKeeper was different to the other algorithms; the 28S gene was more stable than the 18S gene, and both were more stable than the EF1 gene. The reason for this may be because the BestKeeper algorithm uses the original Ct values as input data, so the calculated statistics can indicate false differences.33,34 However, the values for the coefficient of variation (CV) were very close; 0.77 and 0.78, respectively for the different developmental stages, 0.45 and 0.53, respectively for the different nutritional conditions. Therefore, we used values from all four algorithms to rank candidate reference gene stability under all the experimental conditions as a comprehensive indicator of the mean stability of gene expression as did; overall the 18S was still the most stable reference gene in terms of expression.
Through stability analysis, we identified that the 18S rRNA reference gene was suitable for normalization of qPCR data under multiple experimental conditions. Our study was based on qPCR, so the findings will provide direct guidance for future qPCR experiments on P. neoaphidis. Moreover, these findings may also be useful for Northern blot and reverse transcription PCR, techniques that also require reference genes for normalization.8,35,36 It is also possible that, using a combination of two reference genes might further improve the reliability of gene expression by RT-qPCR of P. neoaphidis.
In eukaryotes, the genes most conserved and most widely used are those that encode ribosomal RNA (rRNA).37 These genes encode small subunit ribosomal RNA (18S) and two large subunit ribosomal RNA (28S and 5.8S), in a transcription unit, constituting about 85–90% of total cellular RNA, and are very useful as internal controls.38 Though 28S together with 18S are all processed from a single precursor RNA after transcription from the rRNA cistron, 28S and 18S are synthesized respectively by RNA polymerase I and RNA polymerase II; these are completely different to mRNA synthesis and independent from it. Thus, there are only rare changes in rRNA levels while various conditions affect mRNA expression. As a classic housekeeping gene, the 18S gene has been widely used as a reference for gene expression analysis39; for example in the entomopathogenic fungus Beauveria bassiana,8,32,35 and other species.9,40,41 For this reason we are not surprised that it is also useful for gene expression studies of P. neoaphidis. However, our understanding of P. neoaphidis development and metabolism is still limited, so it is likely that further work on the stability of gene expression in other, as yet not-cloned, reference genes such as GAPDH and β-actin, will also be useful. Furthermore, the use of several reference genes would allow more accurate and reliable normalization of gene expression data.16
ConclusionTo the best of our knowledge, this is the first attempt to select a candidate reference gene for the normalization of RT-qPCR-based gene expression analysis of the entomophthoralean fungus P. neoaphidis. Our results suggest that the 18S gene would be the most reliable for normalizing the expression levels in most sample series. These results provide a foundation for the more accurate and widespread use of RT-qPCR in the analysis of gene expression in P. neoaphidis.
Normalization is a major issue when analyzing gene expression in response to various experimental conditions, and use of incorrect normalization targets could lead to erroneous interpretation of quantitative data. The present study considered the issue of development and nutrition, thus the findings can aid in understanding the mechanisms of development and metabolism. For the issue of fungus/insect interactions, it is more complicated since the condition involves fungal propagules and host tissue. Thus, further experiments are required to assess and validate more reference genes suitable for evaluating fungus/insect interactions.
Conflicts of interestThe authors declare no conflicts of interest.
This study was funded by: the Natural Science Foundation of China (31461143030), the International Foundation for Science (C/4822-1), the Science and Technology Planning Project of Zhejiang Province (2013C37087) and the Zhejiang Natural Science Foundation (LR12C03001).