Humic substances in soil DNA samples can influence the assessment of microbial diversity and community composition. Using multiple steps during or after cell lysis adds expenses, is time-consuming, and causes DNA loss. A pretreatment of soil samples and a single step DNA extraction may improve experimental results. In order to optimize a protocol for obtaining high purity DNA from soil microbiota, five prewashing agents were compared in terms of their efficiency and effectiveness in removing soil contaminants. Residual contaminants were precipitated by adding 0.6mL of 0.5M CaCl2. Four cell lysis methods were applied to test their compatibility with the pretreatment (prewashing+Ca2+ flocculation) and to ultimately identify the optimal cell lysis method for analyzing fungal communities in forest soils. The results showed that pretreatment with TNP+Triton X-100+skim milk (100mM Tris, 100mM Na4P2O7, 1% polyvinylpyrrolidone, 100mM NaCl, 0.05% Triton X-100, 4% skim milk, pH 10.0) removed most soil humic contaminants. When the pretreatment was combined with Ca2+ flocculation, the purity of all soil DNA samples was further improved. DNA samples obtained by the fast glass bead-beating method (MethodFGB) had the highest purity. The resulting DNA was successfully used, without further purification steps, as a template for polymerase chain reaction targeting fungal internal transcribed spacer regions. The results obtained by terminal restriction fragment length polymorphism analysis indicated that the MethodFGB revealed greater fungal diversity and more distinctive community structure compared with the other methods tested. Our study provides a protocol for fungal cell lysis in soil, which is fast, convenient, and effective for analyzing fungal communities in forest soils.
Removal of humic substances from DNA samples is a prerequisite for analyzing soil microbial communities by molecular techniques. Contaminants can be removed before, during, or after cell lysis. To obtain high-quality microbial DNA, a DNA-containing lysate may be purified by adding chemical reagents such as polyvinylpolypyrrolidone (PVPP) and polyethylene glycol or by repeated extraction with phenol–chloroform–isoamyl alcohol during and after cell lysis.1,2 In most cases, however, further purification steps, such as electrophoresis,3 electroelution,4,5 or spin-column chromatography6,7 are needed. Additional steps in DNA extraction and purification are time-consuming and expensive. More importantly, they may result in DNA loss without microbial taxon-specific predilection.8 In other words, DNA loss during extraction and purification is likely to result in underestimation of microbial diversity and misunderstanding of microbial community structure.
Soil pretreatment before cell lysis can minimize the need for additional purification steps. Prewashing of soil with solutions such as 50mM Tris, 20mM ethylenediaminetetraacetic acid (EDTA), 100mM NaCl, and 1% PVPP (hereinafter referred to as TENP) or phosphate-buffered saline (PBS) improves DNA purity,9–11 but trace amounts of humic substances unavoidably remain in DNA samples. Multivalent cations (Ca2+ and Al3+) can be used to precipitate humic substances by chemical flocculation,12–15 however, it is difficult to control the concentration of the cations, and this method can also cause DNA coprecipitation.13,14 Therefore, neither prewashing nor chemical flocculation alone leads to the best performance.
Commercial kits are fast, simple, and effective for soil DNA extraction. The FastDNA® SPIN Kit for Soil (MP Biomedicals, Santa Ana, CA, USA), PowerSoil® DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, USA), and E.Z.N.A.® Soil DNA Kit (Omega Bio-Tek, Inc., Norcross, GA, USA) all use glass beads to rapidly lyse microbial cells. However, kits can be expensive, variable in their performance, and the recipes of the reagents in the kits remain unknown.
Although a large number of studies have compared different soil DNA extraction methods, few have assessed method-related effects on microbial diversity data. Compared with a commercial kit, a modified method (glass beads+lysozyme+proteinase K+freeze-thawing) resulted in more bacterial operational taxonomic units detected.16 Williamson et al.17 demonstrated that among five tested methods, a proteinase K-based method and a commercial kit both resulted in a lower bacterial Shannon–Wiener index. Meanwhile, Zhang et al.18 found that a method using cetyltrimethylammonium bromide (CTAB)–sodium dodecyl sulfate (SDS) had a superior performance in terms of the Shannon–Wiener and Simpson indices of actinobacterial diversity. Significant differences in the resulting microbial diversity data are also observed among commercial kits. Vishnivetskaya et al.19 tested four kits and reported that the FastDNA® SPIN Kit for Soil generated the highest Simpson value, followed by the PowerSoil® Kit and PowerLyzer® Kit, whereas use of the MetaG-Nome® DNA Isolation Kit resulted in the lowest microbial diversity.
Our goal was to improve the fast cell lysis methods used in the kits by determining the optimal prewashing agent and using Ca2+ flocculation to pretreat soil samples prior to cell lysis. Forest soils were used to determine the effectiveness of prewashing agents in removal of soil contaminants because these soils are typically rich in humic substances. In order to evaluate the applicability of soil pretreatment (prewashing+Ca2+ flocculation), three other direct cell lysis methods were assessed. Furthermore, due to their tough cell walls, fungi are generally less sensitive to cell lysis methods. Therefore, terminal restriction fragment length polymorphism (T-RFLP) analysis of fungal communities was used to compare the different cell lysis methods in terms of method-related effects on soil fungal diversity data.
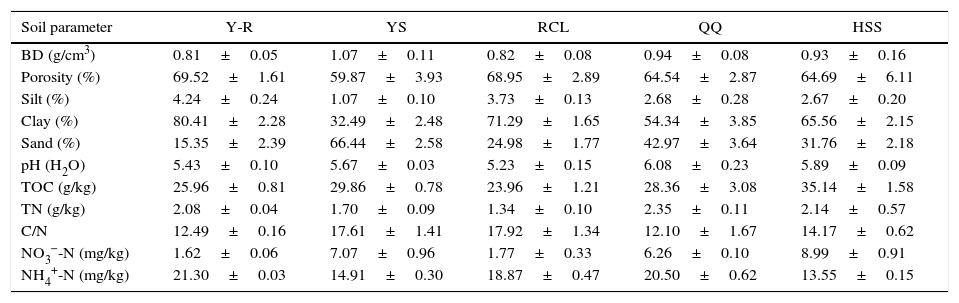
Materials and methodsSoil samplesIn September 2011, soil samples (0–10cm depth) were collected from five forest types in Huoditang located on the south-facing slope of the Qinling Mountains in Shaanxi Province, China. This area is mainly covered by natural secondary forests.20,21 Four sampled forest types were dominated by Chinese pine (Pinus tabulaeformis), sharptooth oak (Quercus aliena var. acuteserrata), Armand pine (Pinus armandii), and Wilson spruce (Picea wilsonii), respectively, while the fifth was a mixed forest type composed of Chinese pine and sharptooth oak. For each forest type, three plots (20m×20m) were established. In each plot, 30 soil cores were collected using a soil corer (3cm in diameter) and pooled into one composite sample. The soil samples were placed in plastic bags and transported to the laboratory on ice. After having been sieved through a 2mm sieve, half of each sample was air dried at room temperature for analysis of soil physical and chemical parameters. This work was conducted in accordance with the Forestry Standards “Observation Methodology for Long-Term Forest Ecosystem Research” of the People's Republic of China (LY/T 1952–2011),22,23 and the soil parameters are presented in Table 1. The other half of each sample was stored in a refrigerator at 4°C until microbial analysis.
Soil physical and chemical properties of the five forest types.
| Soil parameter | Y-R | YS | RCL | HSS | |
|---|---|---|---|---|---|
| BD (g/cm3) | 0.81±0.05 | 1.07±0.11 | 0.82±0.08 | 0.94±0.08 | 0.93±0.16 |
| Porosity (%) | 69.52±1.61 | 59.87±3.93 | 68.95±2.89 | 64.54±2.87 | 64.69±6.11 |
| Silt (%) | 4.24±0.24 | 1.07±0.10 | 3.73±0.13 | 2.68±0.28 | 2.67±0.20 |
| Clay (%) | 80.41±2.28 | 32.49±2.48 | 71.29±1.65 | 54.34±3.85 | 65.56±2.15 |
| Sand (%) | 15.35±2.39 | 66.44±2.58 | 24.98±1.77 | 42.97±3.64 | 31.76±2.18 |
| pH (H2O) | 5.43±0.10 | 5.67±0.03 | 5.23±0.15 | 6.08±0.23 | 5.89±0.09 |
| TOC (g/kg) | 25.96±0.81 | 29.86±0.78 | 23.96±1.21 | 28.36±3.08 | 35.14±1.58 |
| TN (g/kg) | 2.08±0.04 | 1.70±0.09 | 1.34±0.10 | 2.35±0.11 | 2.14±0.57 |
| C/N | 12.49±0.16 | 17.61±1.41 | 17.92±1.34 | 12.10±1.67 | 14.17±0.62 |
| NO3−-N (mg/kg) | 1.62±0.06 | 7.07±0.96 | 1.77±0.33 | 6.26±0.10 | 8.99±0.91 |
| NH4+-N (mg/kg) | 21.30±0.03 | 14.91±0.30 | 18.87±0.47 | 20.50±0.62 | 13.55±0.15 |
YS, Chinese pine; RCL, sharptooth oak; HSS, Armand pine; QQ, Wilson spruce; Y-R, Chinese pine+sharptooth oak; BD, bulk density; TOC, total organic carbon; TN, total nitrogen.
Soil samples from the Armand pine forest were selected for studying contaminant removal because these soils contained the highest amounts of soil organic matter. We used the following five solutions as soil prewashing agents: (1) PBS (137mM NaCl, 2.7mM KCl, 10mM Na2HPO4, 2mM KH2PO4, pH 7.4); (2) 2% NaPO3+1% polyvinylpyrrolidone (PVP), pH 8.5; (3) 0.5M EDTA, pH 8.0; (4) TENP, pH 10.0; and (5) 100mM Tris, 100mM Na4P2O7, 1% PVP, 100mM NaCl, 0.05% Triton X-100, 4% skim milk, pH 10.0 (hereinafter referred to as TNP+Triton X-100+skim milk).24 Briefly, soil samples (0.5g, five replicates per agent) were mixed with 1.5mL of a prewashing agent, followed by vortexing for 3min. The mixture was incubated at 5°C for 5min, centrifuged at 12,000×g for 5min, and the resulting supernatant was collected. Each soil sample was prewashed three times as described, and the supernatant was collected each time. Humic contamination was not quantified but assessed visually.
Ca2+ flocculationOnce the optimal prewashing agent was determined, the prewashed soil samples were treated with 0.6mL of 0.5M CaCl2, and then sterile water was added to a final volume of 2mL. After mixing, the samples were centrifuged (12,000×g) for 10min at 4°C, and the supernatants were discarded. The samples were then subjected to a fast glass bead-beating method (MethodFGB) for cell lysis and extraction with phenol–chloroform–isoamyl alcohol and chloroform–isoamyl alcohol, which is described in detail below. DNA isolated from soil samples that were only prewashed or pretreated (prewashing+Ca2+ flocculation) was compared with that from untreated soils by photography. These three treatments were performed in five replicates each.
Soil DNA extractionBased on the results of the prewashing and Ca2+ flocculation tests, the soil samples from the five forest types were subjected to pretreatment. The soil samples (0.5g) were mixed with 1.5mL of TNP+Triton X-100+skim milk, followed by vortexing and incubation. This prewashing cycle was repeated three times. After final centrifugation, the samples were flocculated with Ca2+, centrifuged, and extracted by one of four cell lysis methods as described below.
MethodFGBFast glass bead beating (FGB) was used in the method. One milliliter of DNA extraction buffer (100mM Tris–HCl, 100mM sodium phosphate, 1.5M NaCl, 1% CTAB, pH 8.0), acid-washed glass beads (0.1mm, 0.4–0.6mm, and 0.8–1.0mm, 0.25g of each type), and 200μL of 20% SDS were added into centrifuge tubes containing the pretreated soil samples. The mixtures were shaken in a MM 400 mixer mill (Retsch, Germany) at 30Hz for 30s three times.
MethodPK25This method required the addition of proteinase K (PK). Prior to cell lysis, 1mL of the DNA extraction buffer and 20μL of proteinase K (10mg/mL) were added into the centrifuge tubes and mixed with the pretreated soil samples, followed by a horizontal oscillation at 250rpm/min for 30min at 37°C. After the oscillation, 200μL of 20% SDS was added, and the samples were incubated at 65°C for 2h with gentle end-over-end inversions every 15min.
MethodSGB26Slow glass bead beating (SGB) was used in the method to lyse cells. The pretreated soil samples were mixed with 0.15g of SDS, the three types of acid-washed glass beads (0.25g of each type), and 1mL of the DNA extraction buffer. The mixtures were incubated at 65°C for 2h with gentle end-over-end inversions every 15min. Then, the tubes were shaken horizontally at 250rpm/min for 30min at 37°C.
MethodLFT27In this method, lysozyme and freeze–thawing (LFT) were used to break microbial cells. The pretreated soil samples were mixed with 1mL of the DNA extraction buffer containing lysozyme (15mg/mL). The mixtures were incubated at 37°C for 2h with gentle end-over-end inversions every 15min, and then 200μL of 20% SDS was added. Three cycles of freezing at −70°C for 30min and thawing at 65°C for 10min were performed to release DNA from microbial cells.
The lysis products obtained by all methods were centrifuged at 8000×g for 15min. The supernatants containing microbial DNA were extracted with an equal volume of phenol–chloroform–isoamyl alcohol (25:24:1, v/v/v), centrifuged at 6000×g at 4°C for 10min, then extracted with chloroform–isoamyl alcohol (24:1, v/v), and centrifuged as before. DNA from the aqueous phase was precipitated with 0.6 volumes of cold isopropanol and 0.1 volumes of 3M sodium acetate (pH 5.2) at room temperature for 2h. Pellets of crude nucleic acids were obtained by centrifugation at 14,800×g for 20min at room temperature, washed with cold 70% ethanol, and dissolved in 100μL of 10mM Tris–HCl buffer (pH 8.0).
Absorbance of the recovered DNA was determined at 230, 260, and 280nm using a NanoDrop 2000 UV-Vis spectrophotometer (Thermo Fisher Scientific, Inc., USA). The device directly displayed DNA concentrations in ng/μL, and these values were converted into μg/g of soil. The purity of the recovered DNA was expressed as A260/A230 and A260/A280 ratios. Agarose gel electrophoresis was used to visually assess the integrity of crude DNA. An aliquot (5μL) of crude DNA was analyzed in a 0.7% (w/v) agarose gel run in 1× Tris–borate buffer at 5V/cm for 1h. The gel was stained with ethidium bromide (0.5μg/mL) and photographed using a Gel Doc XR+ System (Bio-Rad Laboratories, USA).
Polymerase chain reactionThe extracted crude DNA was used as a template for polymerase chain reaction (PCR) without dilution or further purification. The universal fungal primer set (Invitrogen, Inc., Shanghai, China) consisting of ITS1F (5′-CTT GGT CAT TTA GAG GAA GTA A-3′)28 and ITS4 (5′-TCC TCC GCT TAT TGA TAT GC-3′)29 was used for amplification of fungal internal transcribed spacer (ITS) regions. The 5′ end of ITS1F was labeled with the fluorescent dye 6-FAM. PCR reaction mixtures (50μL) contained 2μL of each primer (10μmol/L), 1μL of bovine serum albumin (0.4μg/μL), 25μL of 2× Taq MasterMix (Cowin Biotech, China), 2μL of template DNA, and 18μL of sterilized water. The cycling parameters were 94°C for 5min, followed by 35 cycles of 94°C for 30s, 55°C for 30s, and 72°C for 1min, with a final extension at 72°C for 7min.
T-RFLPPCR amplicons were purified with TIANgel Midi Purification Kit (Tiangen Biotech, China) according to the manufacturer's protocol. Subsequently, the purified amplicons were subjected to restriction endonuclease digestion. Briefly, PCR products were digested with HhaI (GCG^C, Fermentas) at 37°C for 7h to produce terminal restriction fragments (TRFs). The digestion reactions (20μL) contained 4μL of PCR products (0.8–1.0μg), 1μL of 10× buffer, 1μL of the endonuclease (10U) and 14μL of ddH2O. The digestion products were desalted by precipitation with two volumes of cold ethanol and centrifuged at 16,000×g for 15min at 4°C. The DNA pellets were washed twice with 70% cold ethanol and resuspended in 20μL of sterilized ultrapure water.
Prior to capillary electrophoresis, 2μL of digestion products was mixed with 12μL of formamide and 0.5μL of the GeneScan ROX 1000 size standard (Applied Biosystems, USA). The mixtures were denatured at 95°C for 4min and then placed on ice for 5min. The capillary electrophoresis was performed on an ABI 3730xl Genetic Analyzer (Applied Biosystems). The fluorescently labeled 5′-terminal restriction fragments were detected and analyzed by the GeneScan 3.7 software (Applied Biosystems).
Data analysisTRF peaks with a height of less than 50 fluorescence units were excluded, and TRFs of less than 50bp in length were removed. For quality control, the raw data were compiled and uploaded to the T-RFLP analysis EXpedited software–a free web-based tool to aid in the analysis of T-RFLP data.30 Noise filtering was performed for the identification of true peaks by setting the standard deviation multiplier to 1.0. TRFs were then aligned by setting a clustering threshold of 0.5bp. The processed data were imported to MS Excel 2007. Percentages of each TRF peak area relative to the total peak area of each sample were calculated.31 The normalized peak area was defined as relative abundance of each reserved TRF. The Shannon–Wiener (H) and evenness (E) indices were calculated based on the presence/absence and abundance of TRFs, while the richness index (S) was calculated based on the number of TRF. Differences in fungal diversity among the different cell lysis methods were compared using a one-way analysis of variance (ANOVA) with Tukey's post hoc test. The results are shown as the mean±standard deviation (SD). Significant differences were detected at the 0.05 level. The experiment was designed as four cell lysis methods×five forest soils×three sample replicates. Non-metric multidimensional scaling (NMDS) was performed using the Bray–Curtis distance by the PRIMER 5 software.
ResultsSoil prewashingThe effectiveness of prewashing agents in removing soil contaminants was evaluated visually based on the supernatant color (Fig. 1). The results showed that the supernatants of the soil samples treated with TNP+Triton X-100+skim milk had the darkest color (brownish black). TENP and EDTA produced brown supernatants, while those of the samples treated with NaPO3+PVP or PBS were light brown and yellow. Based on the color comparison, TNP+Triton X-100+skim milk extracted larger amounts of soil contaminants than the other agents.
")
Effectiveness of prewashing agents in removal of soil contaminants. Soil samples from the Armand pine forest were prewashed with five agents, and supernatants with different colors were obtained. The dark color indicated substantial extraction of humic contaminants. A, TNP+Triton X-100+skim milk; B, TENP; C, EDTA; D, NaPO3+PVP; E, PBS. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
DNA purity was further improved by calcium chloride flocculation (Fig. 2) using 0.5M Ca2+. DNA isolated from prewashed soil without Ca2+ treatment was light yellow, indicating the presence of trace amounts of humic contaminants. DNA from non-pretreated soil samples was brown, indicating that considerable amounts of humic contaminants were co-extracted with the DNA.

Improvement of soil DNA purity by Ca2+ flocculation. Soil samples from the Armand pine forest were pretreated using three different procedures to obtain crude DNA. The transparent samples indicate high DNA purity. A, crude DNA from non-pretreated soil; B, crude DNA from soil prewashed with TNP+Triton X-100+skim milk; C, crude DNA from soil pretreated using TNP+Triton X-100+skim milk+Ca2+ flocculation.
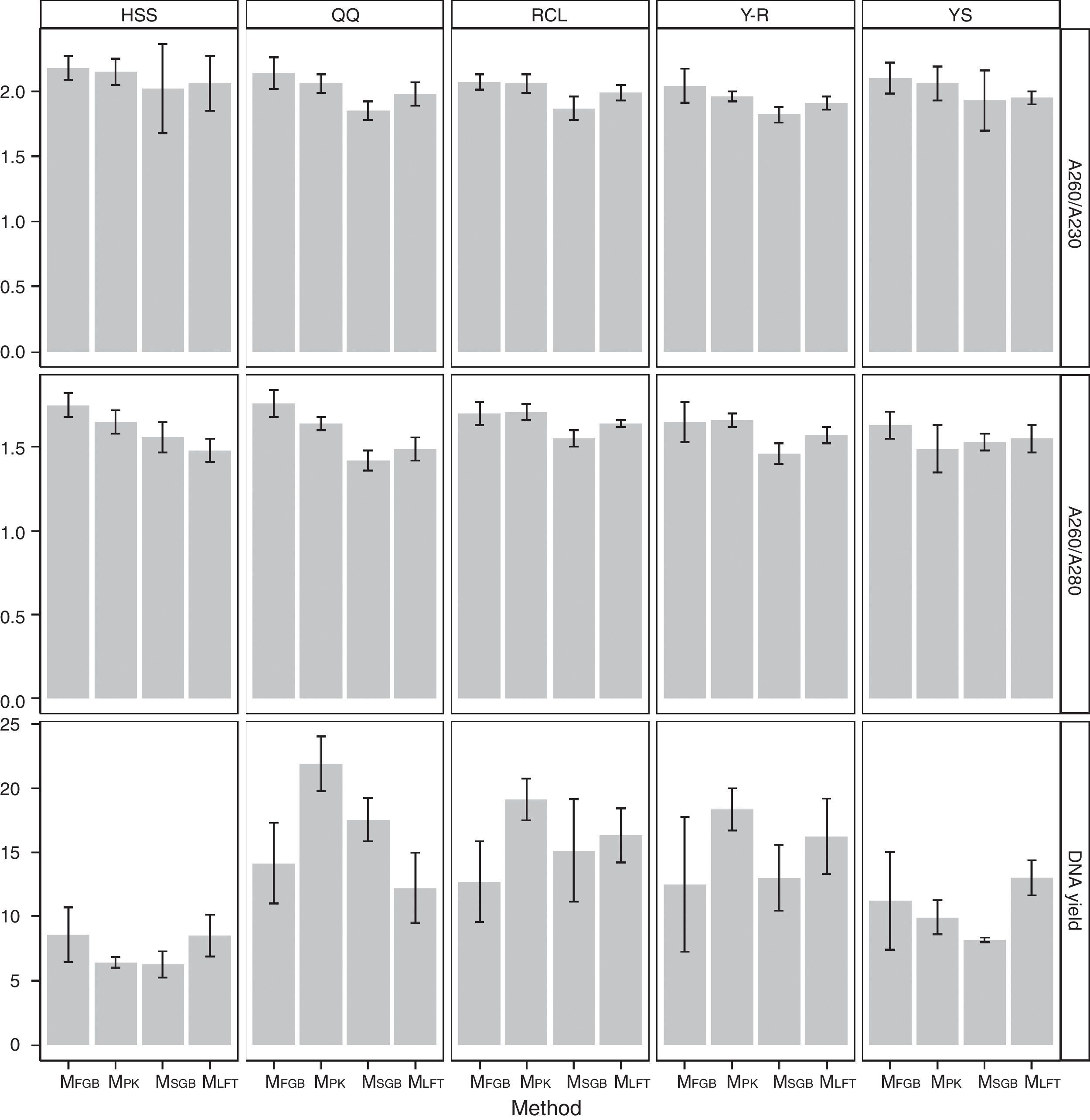
Regardless of the soil origin and cell lysis method, pretreatment resulted in high purity of extracted DNA. The A260/A230 ratios of all DNA samples obtained by the MethodFGB were higher than 2.00, and the A260/A230 ratios of the samples lysed using the other methods were greater than 1.80 (Fig. 3). The A260/A280 ratios of the samples obtained by the MethodFGB were also the highest. Although lower ratios were obtained using the other methods, the ratios were higher than 1.40. However, the MethodFGB was inferior to the other three methods in DNA yield, while the MethodLFT had an excellent yield performance.
PCR
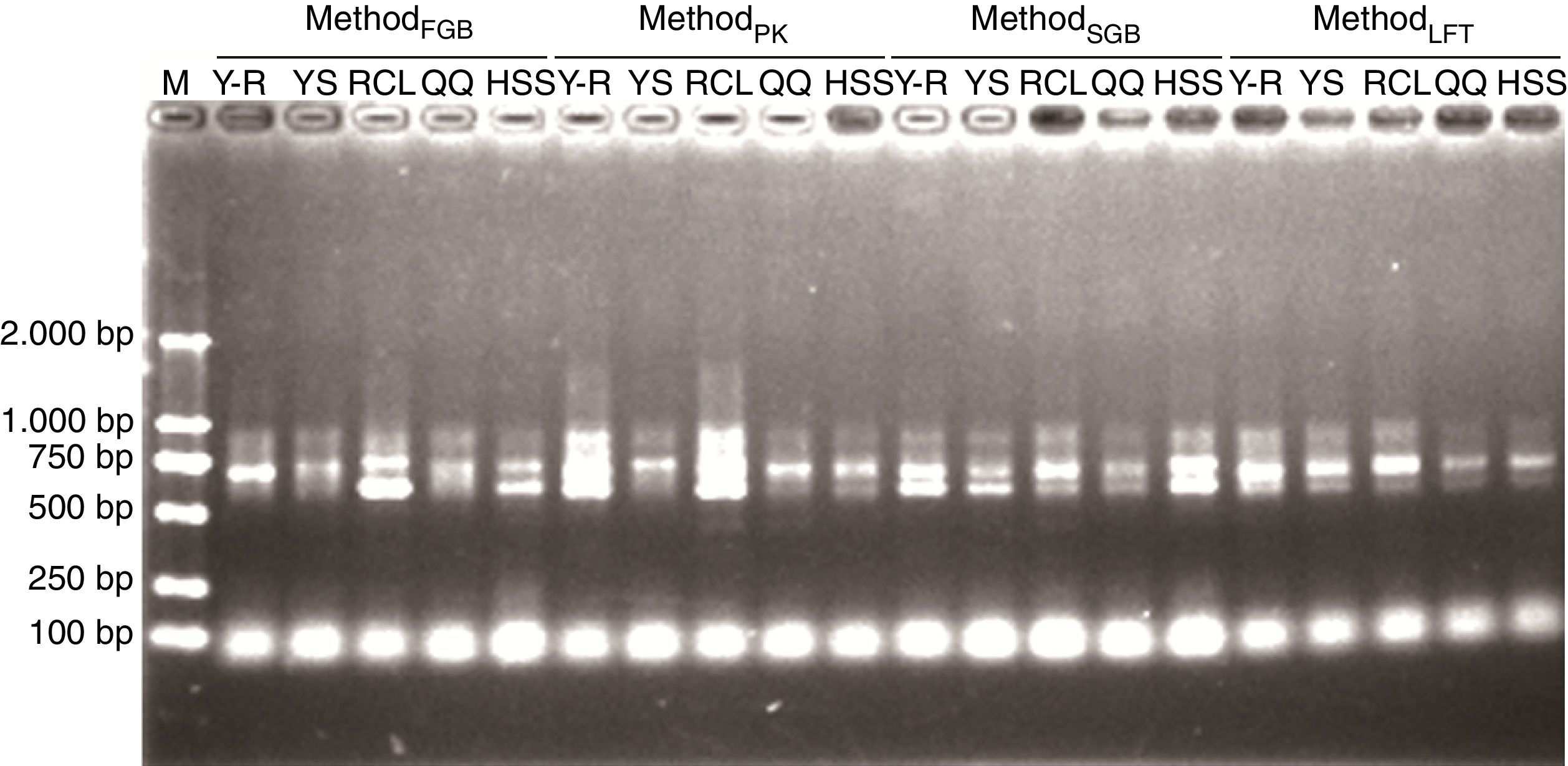
To demonstrate that the pretreatment improves DNA purity, all DNA samples were directly used in PCR. Fig. 4 shows representative DNA samples and indicates that the PCR amplicons are mainly in the size ranging from 500 to 1000 base pairs (bp) and that all the lanes have multiple target bands. The differences among the PCR products in sizes and amounts are consistent with the variability of ITS regions among fungal taxa. Thus, regardless of the soil origin and cell lysis method, the pretreatment made crude DNA available for amplification of target fragments.
T-RFLP
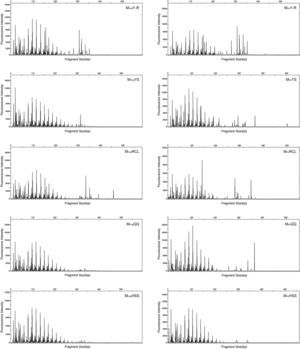
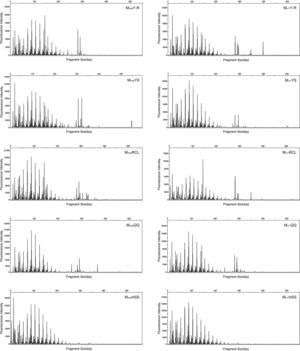
T-RFLP was used to test the practicability of soil pretreatment for analysis of microbial communities. HhaI digestion of the PCR products showed that most TRF peaks were below 440bp (Figs. S1 and S2). The TRF numbers and fluorescence intensity differed among the profiles, indicating that the samples represented fungal communities that differed in diversity and species composition.
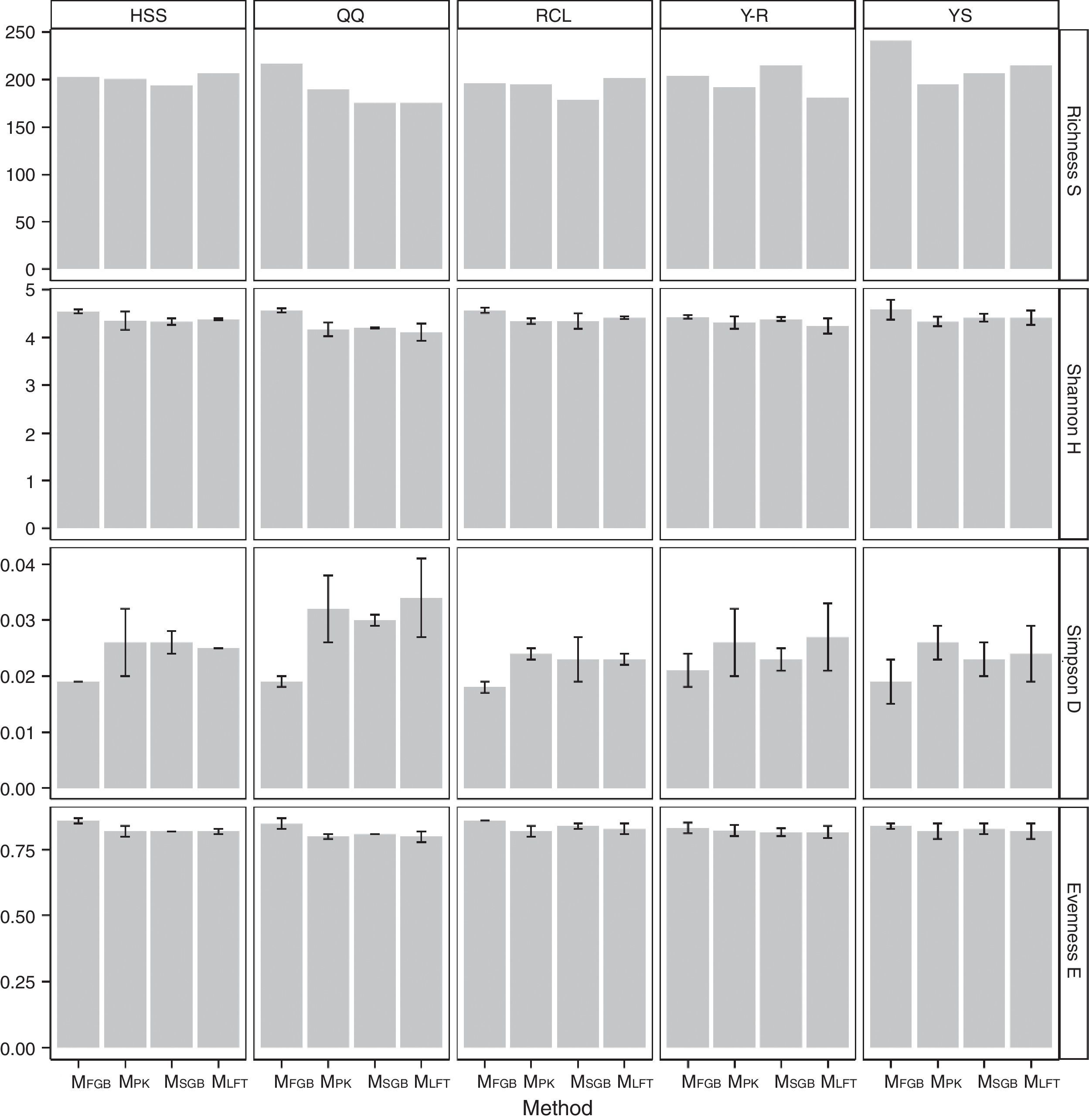
Fungal diversity indices were determined from the T-RFLP profiles to reveal the effects of the cell lysis methods (Fig. 5). The richness (S) index indicated that DNA extracted by the MethodFGB produced the largest numbers of TRFs in the cases of the YS and QQ soils, and relatively high S values were obtained for the DNA samples from the other forest soils. In the cases of the RCL and HSS soils, the maximal numbers of TRFs were produced when using the DNA samples extracted by the MethodSGB. In the cases of the Y-R soil samples, DNA extracted by the MethodPK had the highest value of S. For all the soils, DNA extracted by the MethodFGB had the highest Shannon (H) and evenness (E) indices. Generally, a low Simpson (D) index indicates a high fungal diversity, and in the present study, the lowest D indices were obtained when using MethodFGB DNA samples. Furthermore, in most cases the three diversity indices, Shannon (H), Simpson (D), and evenness (E), reached significant levels of difference (p<0.05 or p<0.01) for MethodFGB DNA samples compared with those extracted by the other methods. Overall, the use of the MethodFGB resulted in the greatest estimates of fungal diversity.

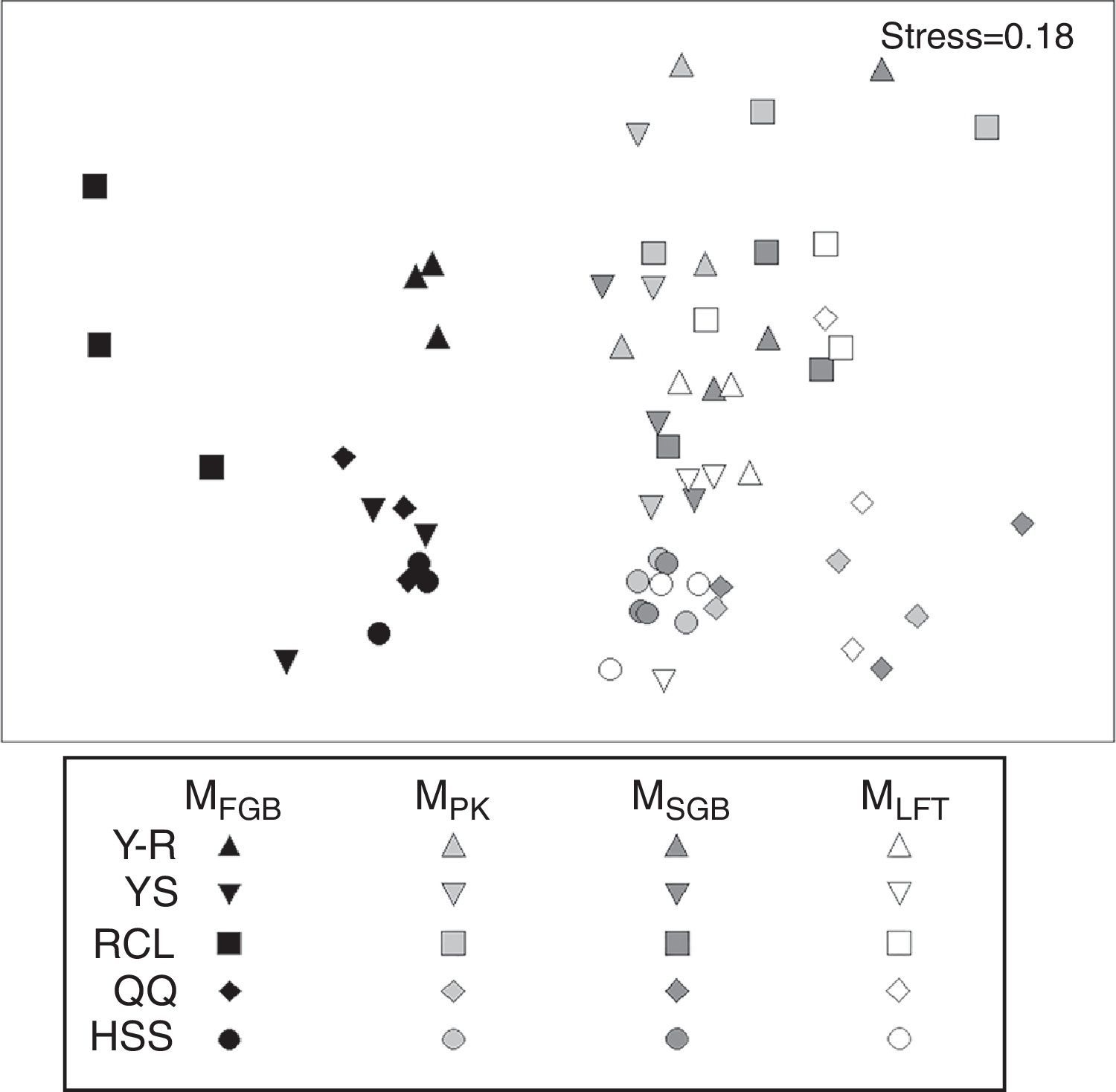
NMDS analysis showed differences among the tested cell lysis methods (Fig. 6). All sample points for the MethodFGB are distributed on the left side of the NMDS plot, while those for the other methods are scattered on the other side. The points representing the YS, HSS, and QQ soils treated by the MethodFGB are closely clustered, indicating that these three forest types had similar soil fungal communities. The Y-R and RCL soils had distinct community compositions. The points for the other methods are intermixed so that no clear between- or within-method tendency is observed.
. The colors and shapes of the dots represent different cell lysis methods and forest soils, respectively. MFGB, MethodFGB; MPK, MethodPK; MSGB, MethodSGB, MLFT, MethodLFT; YS, Chinese pine; RCL, sharptooth oak; HSS, Armand pine; QQ, Wilson spruce; Y-R, Chinese pine+sharptooth oak.")
NMDS analysis of fungal T-RFLP profiles obtained using all cell lysis methods and forest soils (n=3). The colors and shapes of the dots represent different cell lysis methods and forest soils, respectively. MFGB, MethodFGB; MPK, MethodPK; MSGB, MethodSGB, MLFT, MethodLFT; YS, Chinese pine; RCL, sharptooth oak; HSS, Armand pine; QQ, Wilson spruce; Y-R, Chinese pine+sharptooth oak.
Elimination of humic contaminants during DNA extraction has been an important focus of research because contaminants can inhibit downstream applications.32 Humic contaminants can produce covalent complexes between humic acid and DNA or proteins.33 Phenol groups of humic acid may combine with amino groups, leading to denaturation of biological macromolecules or quinone formation. Additionally, humic contaminants may chelate Mg2+, repressing DNA polymerase activity.32,34 PCR may be completely blocked by 10ng of contaminants.35,36
Soil pretreatment prior to cell lysis prevents co-extraction of DNA and fuscin or heavy metals.13 We used several common buffers to prewash soils, including NaPO3+PVP or PBS, but in our study they showed a poor performance, leading to the formation of light brown or light yellow supernatants, respectively. PVP can form insoluble complexes with soil polyphenols and also combine with polysaccharides.37 The colors of TENP and EDTA supernatants were dark brown, indicating that more humus was extracted.
TENP has been extensively used to remove soil contaminants.9,11 Among the TENP components, Tris is considered an excellent buffer providing a stable buffering environment, while EDTA is a chelating agent playing a major role in the removal of heavy metals from soils and protecting DNA from DNase degradation.38 The addition of Triton X-100 and skim milk to TENP further facilitated the extraction of humic contaminants as indicated by the formation of black brown and almost completely opaque supernatants. Triton X-100 may enhance DNA water solubility, degrade carbohydrates, and alleviate the adhesion of microbial cells and carbohydrates.39,40 Skim milk competes with DNA-adsorbing soil particles, thus increasing the effectiveness of extraction,41 and may also adsorb humus. Overall, Triton X-100 and skim milk were important for the overall performance of TNP+Triton X-100+skim milk and made the solution superior to TENP and the other agents tested in the present study. Soil prewashing with TNP+Triton X-100+skim milk did not solve all DNA extraction problems, there still were traces of contaminants in the crude DNA after three prewashing cycles. Although clear supernatants could be collected after prewashing for six times with this agent, this would make DNA extraction time-consuming.
Multivalent cations neutralize negatively charged sites on humic substances, subsequently forming precipitating multivalent cationic complexes.42 Humic substances contain many carboxyl and hydroxyl groups, and their physical–chemical characteristics are similar to those of the phosphate groups of the sugar–phosphate backbone of DNA, therefore, DNA is also flocculated with multivalent cations in a similar manner.42 Ernst et al.14 suggested that the Ca2+ concentration in a DNA extraction buffer should not exceed 4%. Braid et al.12 and Dong et al.13 found that in contrast to Ca2+, Al3+ performed better in the removal of contaminants, however, the DNA yield decreased with an increased Al3+ concentration. Li et al.15 reported that precipitation of contaminants with 1mL of 0.5M CaCl2, followed by sodium oxalate addition for the removal of excess Ca2+, improved soil DNA purity. In this study, the volume of 0.5M Ca2+ was decreased to 0.6mL to reduce DNA precipitation and maintain high DNA purity. This low volume should be used with TNP+Triton X-100+skim milk.
In this study, the A260/A230 ratios of all DNA samples were higher than 1.80, and some were even greater than 2.0, especially those of the samples extracted by the MethodFGB and MethodLFT. The ratios for the MethodPK and MethodSGB ranged from 1.80 to 1.90, also meeting the requirements for PCR. The A260/A280 ratios of all DNA samples failed to show the desired 1.80 value, but the DNA samples obtained by the MethodFGB had values much closer to the threshold. The purity of DNA extracted by the MethodFGB can be comparable to that of DNA obtained with commercial kits.43–45 The DNA samples isolated using the other methods had higher or lower levels of protein contamination, but the PCR results indicated that the A260/A280 ratios were acceptable. Based on these results, the pretreatment (prewashing+Ca2+ flocculation) is suitable for a range of cell lysis methods and soil samples.
Normally, PCR is used to test inhibitory actions of contaminants present in environmental DNA samples. In the present study, the crude DNA samples from the pretreated soils were not subjected to extra purification or dilution steps but were instead directly used as templates for PCR. Multiple target fragments were amplified, indicating that the most inhibitory humic contaminants were removed by the pretreatment. The results also indicated that the pretreatment was compatible with subsequent DNA manipulations. In practice, appropriate dilution of crude DNA preparations may overcome the inhibition of PCR caused by excess template and residual proteins, resulting in better PCR performance.
Cell lysis methodAssessment of microbial diversity and community structure using T-RFLP is based on the assumption that DNA samples contain the vast majority of microbial information. T-RFLP profiles consist of many peaks representing different TRFs. The area or height of each peak is considered to represent the TRF abundance.46–48 In the present study, the T-RFLP profiles differed in the TRF numbers, peak areas, and heights, indicating differences among soil fungal communities in the distinct forest types. Therefore, the PCR products obtained after soil pretreatment, which was combined with different DNA extraction methods, could be used for T-RFLP analyses of soil fungal communities of the different forest types. The T-RFLP profiles from the same forest type, obtained using DNA extracted by different methods, produced similar TRF patterns, reflecting high stability of the communities, regardless of the method used.
In this study, four cell lysis methods were used for fungal cell lysis. Glass bead beating is recognized as an excellent way to break thick fungal cell walls.8,49,50 Both MethodFGB and MethodSGB used glass beads to break cells, but the former eventually revealed higher fungal diversity than the latter. We speculated that despite the shorter beating time used in the MethodFGB, there were more collisions between the glass beads and fungal cells at a frequency of 30Hz/s. It is possible that more violent beating applied for a shorter period of time breaks cells more efficiently than that with a lower frequency applied for a longer time. The MethodSGB had a low efficiency of breaking cells due to its gentleness. The performance of the MethodLFT was unsatisfactory in terms of both fungal diversity and NMDS data. Repeated freezing and thawing might have broken cells that mostly belonged to dominant populations or to species with fragile cell walls in the communities, resulting in incomplete cell lysis. He et al.10 found that bead beating was superior to high salt+proteinase K in fungal cell lysis, which is consistent with our results, perhaps, gentle lysis is ineffective in breaking fungal cell walls.49
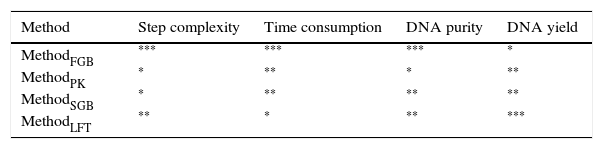
It is usually assumed that a high DNA yield indicates that most of the genetic diversity of a soil microbial community has been sampled, but this may not always be the case. Community structure is evaluated based on microbial diversity, while community similarity is based on shared species, a larger number of shared species reveals greater similarity between communities. In this study, the most efficient cell lysis method (MethodFGB) produced diversity estimates that were higher than those obtained by the other methods, thus leading to the conclusion that the soil communities were distinct. This level of resolution could not be attained using the other methods.The four cell lysis methods tested in our study were comprehensively assessed based on the ease of use and the results (Table 2). The MethodFGB extracted the purest DNA, although the total DNA yield was lower than those obtained by the other methods. In practice, high DNA purity is often more important and more difficult to obtain than a high DNA yield.51 For instance, a picogram of a highly purified DNA template may be sufficient for a successful PCR. The MethodFGB required fewer cell lysis steps and less time for bead beating to break fungal cells than the other methods. The full MethodFGB protocol took 3h, while the other methods needed 5.5 to 7h. Furthermore, the MethodFGB was better than the other cell lysis methods in terms of both fungal diversity and community composition. Hence, combined with soil pretreatment, the MethodFGB was the best cell lysis method among those that we tested. The combination of pretreatment and fast bead beating in a single protocol for fungal DNA extraction from soil resulted in a method that is fast, convenient, and effective for analyzing fungal communities of forest soils.
ConclusionsTo achieve highly effective and economical soil DNA extraction, we performed a series of tests. Ultimately, the combination of TNP+Triton X-100+skim milk, Ca2+ flocculation, and MethodFGB were identified to have the optimal performance. Below, we summarize the entire procedure so that it can be applied to soil DNA extraction.
PrewashingSoil samples (0.5g) were mixed with 1.5mL of TNP+Triton X-100+skim milk (100mM Tris, 100mM Na4P2O7, 1% PVP, 100mM NaCl, 0.05% Triton X-100, and 4% skim milk, pH 10.0), followed by vortexing for 3min. The mixtures were incubated at 55°C for 5min, centrifuged at 12,000×g for 5min, and the supernatants were discarded. This prewashing cycle was performed three times.
Ca2+ flocculationThe samples were mixed with 0.6mL of 0.5M CaCl2, and sterile water was added to a final volume of 2mL. Then, the mixtures were centrifuged at 12,000×g for 10min at 4°C, and the supernatants were discarded.
DNA extractionSoil microbial DNA was extracted using the MethodFGB as follows. One milliliter of DNA extraction buffer (100mM Tris–HCl, 100mM sodium phosphate, 1.5M NaCl, 1% CTAB, pH 8.0), acid-washed glass beads (<0.1mm, 0.4–0.6mm, and 0.8–1.0mm, 0.25g of each type), and 200μL of 20% SDS were added to the samples. The mixtures were shaken vigorously in a RETSCH MM 400 Mixer Mill at 30Hz for 30s three times, followed by centrifugation at 8000×g for 15min.
Supernatants containing DNA were recovered and successively extracted with an equal volume of phenol–chloroform–isoamyl alcohol (25:24:1, v/v/v) and chloroform–isoamyl alcohol (24:1, v/v). The resulting aqueous phase was recovered by centrifugation at 6000×g at 4°C for 10min, and DNA was precipitated with 0.6 volumes of cold isopropanol and 0.1 volumes of 3M sodium acetate (pH 5.2) at room temperature for 2h. DNA pellets were obtained by centrifugation at 14,800×g for 20min at 4°C, washed twice with cold 70% ethanol, and resuspended in 100μL of 10mM Tris–HCl buffer (pH 8.0).
Conflicts of interestThe authors declare no conflicts of interest.
DisclaimerMention of a trademark, proprietary product, or vendor does not constitute a guarantee or warranty of the product by the U.S. Dept. of Agriculture and does not imply its approval or the exclusion of other products or vendors that may also be suitable.
This work was funded by the programs “Technical management system for increasing the capacity of carbon sink and water regulation of mountain forests in the Qinling Mountains” (201004036) and “Detecting, simulating and applying techniques for coupling of carbon, nitrogen and water in forest ecosystems” (201104009), approved by the State Forestry Administration of China. This work was also supported by CFERN & GENE Award Funds on Ecological Paper.