







El síndrome de Marfan (SM) es una afectación hereditaria multisistémica del tejido conectivo con gran heterogeneidad fenotípica, por las más de 500 mutaciones implicadas reconocidas y también por la gran variabilidad posible en la expresión de una misma mutación del gen responsable de codificar la fibrilina 1 (FBN1). A pesar del favorable impacto en el pronóstico de avances introducidos en las últimas décadas, las complicaciones cardiovasculares continúan siendo la principal causa de muerte en el SM. Dada la elevada mortalidad prematura, en pacientes con SM no tratados es fundamental instaurar medidas tempranas para aumentar su esperanza de vida.
En esta revisión se expondrán recomendaciones prácticas extraídas de Guías relativas al manejo de las manifestaciones cardiovasculares en el SM, desde el diagnóstico y el seguimiento hasta el tratamiento, basadas en información obtenida de estudios de seguimiento, ensayos aleatorizados y modelos experimentales1-3.
Afectación cardiovascular en el SMLas manifestaciones cardiovasculares del SM pueden afectar al corazón, entre las que destaca la afectación mitral, o a los grandes vasos, la arteria pulmonar y la aorta; la patología aórtica es la que presenta mayor impacto pronóstico3. La enfermedad aórtica está presente en el 60-80% de los adultos con SM y, hasta la introducción de la cirugía profiláctica de la aorta proximal, sus complicaciones eran responsables de hasta el 90% de la mortalidad. La afectación típica de la aorta en el SM consiste en la progresiva dilatación de la raíz, comenzando a nivel de los senos de Valsalva, aunque puede extenderse a otros segmentos. El aneurisma de aorta ascendente puede ocasionar disección, rotura aórtica aguda o insuficiencia aórtica (IAo).
En la histopatología, los segmentos aórticos afectados presentan degeneración de la capa media parietal, con fragmentación y desorientación de fibras que finalmente conlleva la pérdida de la lámina elástica y su reemplazo por una matriz desorganizada de proteoglucanos. Las regiones de degeneración y la escasez de células dan una imagen lacunar, conocida como necrosis quística de la media, que no es ni necrosis, ni quística ni patognomónica del SM, y que puede presentarse en otras enfermedades degenerativas como la hipertensión arterial (HTA). La continua destrucción de las fibras elásticas de la media aumenta progresivamente la rigidez aórtica, facilitando la dilatación y el adelgazamiento de la pared. Todo ello aumenta la tensión parietal y facilita, aún más, el daño de fuerzas de torsión y cizallamiento que actúan sobre las capas íntima y media de la aorta a lo largo del ciclo cardiaco. El estrés hemodinámico, que provoca daño parietal aórtico incluso en tejido normal, en el SM exacerba la elastólisis exagerada debida a metaloproteasas, y la disregulación del factor de crecimiento β transformador de citoquinas (TGF-β) que promueve la apoptosis de células musculares lisas vasculares. El resultado final es un círculo vicioso que acelera la degeneración parietal y provoca dilatación aórtica progresiva, que se intensificará por estados de sobrecarga de presión como la HTA, o de volumen, como la IAo2.
Manejo de la afectación aórticaDe forma resumida, el manejo de la patología de la aorta en el SM consiste en:
- •
Estudio clínico y mediante técnicas de imagen para detectar y cuantificar la progresión de la dilatación aórtica.
- •
Terapia farmacológica y otras medidas dirigidas a retrasar la dilatación y prevenir las complicaciones.
- •
Cirugía electiva de la aorta cuando su dilatación suponga un riesgo elevado o exista regurgitación relevante.
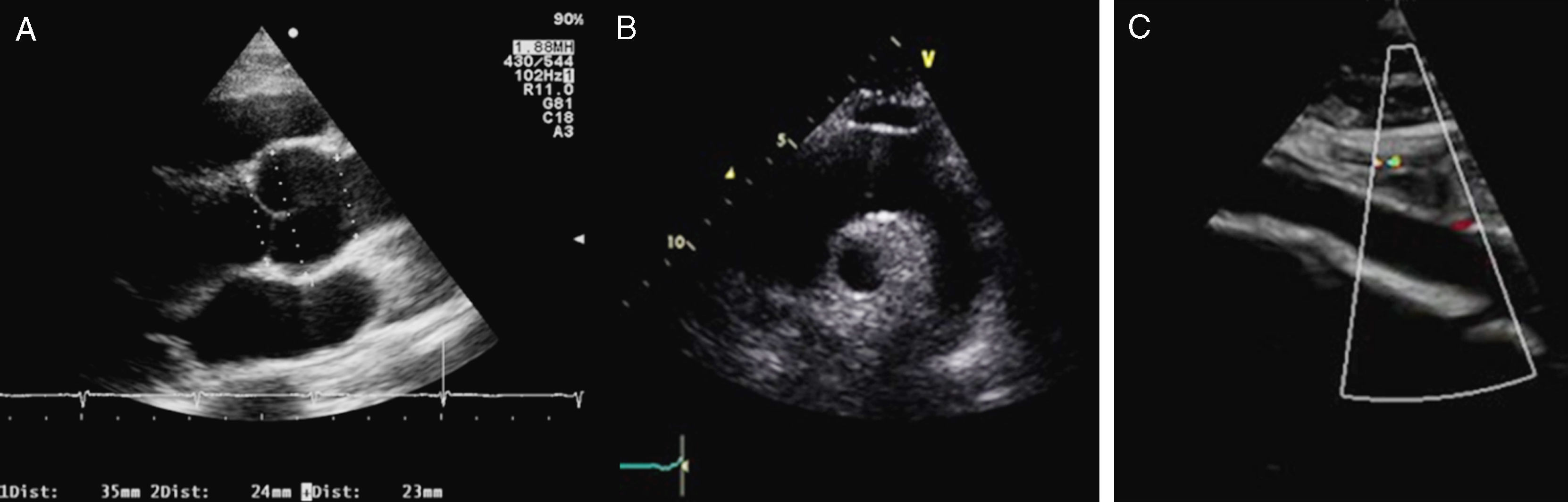
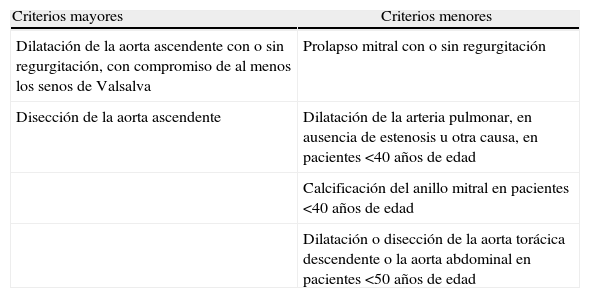
La evaluación clínica inicial de todo paciente con sospecha de SM debe incluir anamnesis y examen físico completos. El diagnóstico de certeza se puede alcanzar casi en el 90% de los casos mediante la clasificación nosológica de Gante2; el empleo de estudios genéticos y el diagnóstico diferencial se discutirán en otro artículo de esta monografía. Para completar la información sobre criterios diagnósticos (tabla 1) se realizará una técnica de imagen que permita evaluar la aorta ascendente y las válvulas cardíacas. El ecocardiograma transtorácico (ETT) representa la principal técnica para el diagnóstico del compromiso cardiovascular en la evaluación inicial, permitiendo explorar la raíz aórtica, la aorta ascendente proximal y el cayado (fig. 1). Los diámetros máximos del anillo, de los senos de Valsalva, de la unión sinotubular y de la aorta ascendente deben ser medidos perpendicularmente al eje longitudinal de la aorta (fig. 2). Los datos obtenidos serán comparados en nomogramas con los valores esperados según la edad, el sexo y la superficie corporal (SC)1. La severidad de la afectación aórtica se relaciona con el grado y la extensión de la dilatación, siendo mayor cuando se extiende desde la raíz por la aorta ascendente hasta el cayado. El segundo ETT se realizará a los 6 meses del diagnóstico para determinar la velocidad de crecimiento. Si el diámetro permanece estable, el ETT puede realizarse anualmente, pero si se detecta dilatación acelerada o cuando se aproxima a 45mm, la evaluación deberá ser más frecuente (tabla 2).
Criterios de Gante: afectación cardiovascular en el síndrome de Marfan
| Criterios mayores | Criterios menores |
| Dilatación de la aorta ascendente con o sin regurgitación, con compromiso de al menos los senos de Valsalva | Prolapso mitral con o sin regurgitación |
| Disección de la aorta ascendente | Dilatación de la arteria pulmonar, en ausencia de estenosis u otra causa, en pacientes <40 años de edad |
| Calcificación del anillo mitral en pacientes <40 años de edad | |
| Dilatación o disección de la aorta torácica descendente o la aorta abdominal en pacientes <50 años de edad |
 El eje paraesternal largo muestra la raíz aórtica y la aorta ascendente y sus diámetros. B) El eje supraesternal muestra la aorta ascendente, el cayado y la aorta torácica descendente proximal. C) Aorta abdominal.")
, proyección paraesternal. Se muestran los niveles de medición de la aorta proximal.")
Seguimiento cardiovascular en el síndrome de Marfan
| Anamnesis, examen físico, ecocardiograma: |
| • Al inicio y a los 6 mesesa |
| • Posteriormente: cada año, si el ritmo de crecimiento es estable y sin complicacionesa |
| TC o RM: |
| • Si hay dilatación o disección aórticas. |
| • Tras la cirugía, antes del alta, a los 6 meses, y luego anualmente. |
| La evaluación será más frecuente a medida que la raíz aórtica se acerque a 45mm o si se registra una tasa de crecimiento acelerada (>5 mm/año) |
aRecomendaciones clase I, nivel de evidencia C.
bSe considera de utilidad corregir los diámetros aórticos de acuerdo a la edad y el tamaño corporal (clase IIa, nivel de evidencia C).
Adaptada de Hiratzka et al1.
A pesar de que la ETT es la técnica más utilizada para monitorizar el tamaño de la raíz aórtica, su precisión depende del operador. La tomografía computarizada (TC) o la resonancia magnética (RM) son más precisas y deben ser usadas si el ETT no da una imagen adecuada de la aorta. Es conveniente saber que las medidas ecocardiográficas, al realizarse entre bordes internos, pueden ser hasta 4mm inferiores a las obtenidas con RM o TC, en las que se incorpora el grosor de la pared.
Prevención de complicaciones cardiovascularesTratamiento farmacológicoBetabloqueantesDiversos ensayos clínicos han demostrado que, al disminuir el estrés hemodinámico en la aorta proximal, los betabloqueantes, especialmente si se emplean desde etapas precoces de la enfermedad, pueden enlentecer el ritmo de dilatación de la aorta y retrasar el momento de aparición de las complicaciones aórticas en el SM (IAo, disección, necesidad de cirugía; insuficiencia cardíaca congestiva; o muerte). Estos beneficios se han observado en todos los grupos de edad, aunque son más marcados en pacientes con dilatación aórtica no severa. Actualmente las guías recomiendan usar betabloqueantes en dosis adecuadas en todos los pacientes con SM que los toleren, independientemente del grado de dilatación aórtica. Dado que la velocidad de crecimiento de la aorta cambia a lo largo de la vida, presentando un pico prepuberal, se recomienda iniciar el tratamiento en la infancia y mantenerlo de por vida, incluso en pacientes sometidos a cirugía profiláctica de la aorta. Los efectos del tratamiento farmacológico deben evaluarse periódicamente para asegurar un manejo óptimo de la FC y de la PA (tabla 3).
Tratamiento farmacológico en el síndrome de Marfan
| Betabloqueantes | Usar siempre en SM, salvo en casos de intoleranciaaAtenolol: más utilizado (larga vida media y cardioselectividad) |
| Dosis: titular hasta FC en reposo <60 lpm y <100 lpm en ejercicio, si la PA lo permite. Monitorizar la eficacia y las dosis en visitas periódicas | |
| Antagonistas del calcio | Verapamilo: fármaco de segunda línea en pacientes que no toleran el betabloqueo |
| IECA | Asociados a betabloqueante cuando se necesite medicación adicional para controlar la PA, especialmente aquellos con disección crónica |
| ARA II | Bloqueo AT1 (losartán) asociado a betabloqueo; en ensayos pequeños no aleatorizados, mayor eficacia en retrasar el ritmo de crecimiento aórticob. |
| Bloqueo AT2, asociado a betabloqueo; uso alternativo a IECA cuando se necesite medicación adicional para el control de PA |
La influencia del sistema renina-angiotensina (SRA) en la degeneración parietal aórtica del SM parece cada vez más importante. La angiotensina II (ATII) estimula la expresión de metaloproteasas y promueve la apoptosis de células musculares lisas de la pared aórtica. Los modelos experimentales han demostrado que la deficiencia de FBN1 aumenta el TGF-β activo, causando detención del ciclo de diferenciación celular, aumento de la apoptosis y depósito de matriz extracelular. El bloqueo del SRA, mediante inhibidores de la enzima de conversión de la angiotensina (IECA) o con antagonistas de los receptores de la angiotensina II (ARAII), produce efectos beneficiosos a distintos niveles. Los IECA contribuyen, además del control de la PA, a la disminución de la rigidez de la pared aórtica. El bloqueo selectivo del receptor tipo 1 (AT1) de la ATII podría aminorar los efectos deletéreos del TGF-β, independientemente de sus efectos sobre el control de la PA. Aunque en modelos murinos el losartán ha demostrado detener e incluso revertir manifestaciones del SM, incluyendo el aneurisma aórtico y sus complicaciones, se está a la espera de los resultados de ensayos controlados en humanos que actualmente están en curso.
Es importante insistir que el tratamiento médico, basado fundamentalmente en el beta-bloqueo, al que se puede asociar el bloqueo del SRA, sirve para retrasar la dilatación aórtica3, pero ningún fármaco, hasta el momento, ha demostrado prevenir el desarrollo de disección ni evitar la necesidad de cirugía en humanos.
Actividad físicaPara reducir el estrés hemodinámico en el SM, la restricción de la actividad física complementa la terapia farmacológica. El ejercicio isométrico intenso está contraindicado debido a los aumentos marcados de la PA periférica y al estrés de la pared aórtica proximal2. También están contraindicados los deportes de competición, los de contacto y los que incluyen cambios marcados en la presión atmosférica, para prevenir el traumatismo arterial y el neumotórax. Dado que el ejercicio dinámico se asocia con menor estrés aórtico, por la disminución de la resistencia vascular periférica y de la PA diastólica, en pacientes sin riesgo elevado se considera segura la práctica de actividad aeróbica hasta de moderada intensidad (tabla 4).
Recomendaciones para la actividad física en el síndrome de Marfan
| Tipo de paciente | Recomendación |
| Todo paciente con SMCualquier grado de dilatación de raíz aórtica | Evitar deportes de contacto y con riesgo de impacto corporal |
| Bajo riesgo: todos los siguientesSin dilatación de raíz aórtica | Actividad estática y dinámica de intensidad baja y moderadaa |
| • Adultos, raíz <40 mm | |
| • Niños y adolescentes: raíz Z-score <2 | |
| Insuficiencia mitral menor que moderada | |
| Sin antecedentes familiares de riesgo: | |
| • Disección o muerte súbita | |
| Riesgo: alguno de los siguientes | Solo aconsejable actividad dinámica de baja intensidad |
| Dilatación de raíz aórtica | |
| • Adultos, raíz ≥40 mm | |
| • Niños y adolescentes: raíz Z-score ≥2 | |
| Insuficiencia mitral moderada o severa | |
| Cirugía previa de raíz aórtica | |
| Disección crónica | |
| Antecedentes familiares de: | |
| • Disección o muerte súbita | |
El tratamiento betabloqueante se considera estándar para todos los pacientes.
aFC máxima durante actividad <100 lpm (adultos) y hasta 110 lpm (niños) con betabloqueo.
bSi hay práctica deportiva habitual, es conveniente el seguimiento del ritmo de crecimiento de la raíz aórtica mediante ETT semestral.
La presencia de insuficiencia aórtica importante con dilatación de la raíz hace desaconsejable cualquier tipo de práctica deportiva.
Basada en Maron et al. J Am Coll Cardiol. 2005;45:1340-1345.
En el SM se recomienda cirugía profiláctica de la raíz aórtica y de la aorta ascendente, por la elevada mortalidad del reemplazo aórtico de emergencia y porque la disección tipo A y la rotura aórtica son las complicaciones con mayor impacto en la supervivencia. Aunque técnicamente más complejas, las técnicas de conservación valvular, remodelado o reimplantación (fig. 3) suelen preferirse en general a los tubos valvulados, siempre que se ofrezcan buenos resultados3–6.
 Dilatación de la raíz aórtica con incompetencia valvular. B) Resultado tras la cirugía de reimplantación de la aorta (técnica de David).")
Dado que el riesgo de disección y la mortalidad son proporcionales al tamaño de la aorta proximal, las guías recomiendan cirugía electiva en adultos cuando el diámetro externo es ≥50mm. La cirugía también debe ser considerada en pacientes con diámetro <50mm y que presenten factores de riesgo adicional: a) rápido crecimiento del diámetro aórtico (>5mm/año); b) antecedente familiar de disección o rotura aórtica, y c) presencia de IAo significativa (tabla 5).
Criterios para cirugía electiva de la aorta proximal en adultos con síndrome de Marfan
| Recomendaciones clase I, nivel de evidencia C |
| Diámetro externo de aorta proximal ≥ 50 mm |
| <50mm con alguno de los siguientes factores de riesgo: |
| • Antecedente familiar de disección o de rotura aórtica |
| • Rápida progresión del diámetro aórtico (>5 mm/año) |
| • Insuficiencia aórtica significativa (moderada o mayor) |
| Recomendaciones clase IIa, nivel de evidencia C |
| En mujeres con SM que deseen el embarazo parece razonable reemplazo de raíz y aorta ascendente cuando el diámetro >40 mm |
| Se recomendará cirugía aórtica cuando el cociente del área máxima aórtica proximal (en cm2) dividido por la estatura en metros sea superior a 10, ya que los pacientes más bajos y el 15% de los pacientes con SM tienen disección aórtica con diámetros <50 mm |
Adaptada de Hiratzka et al1.
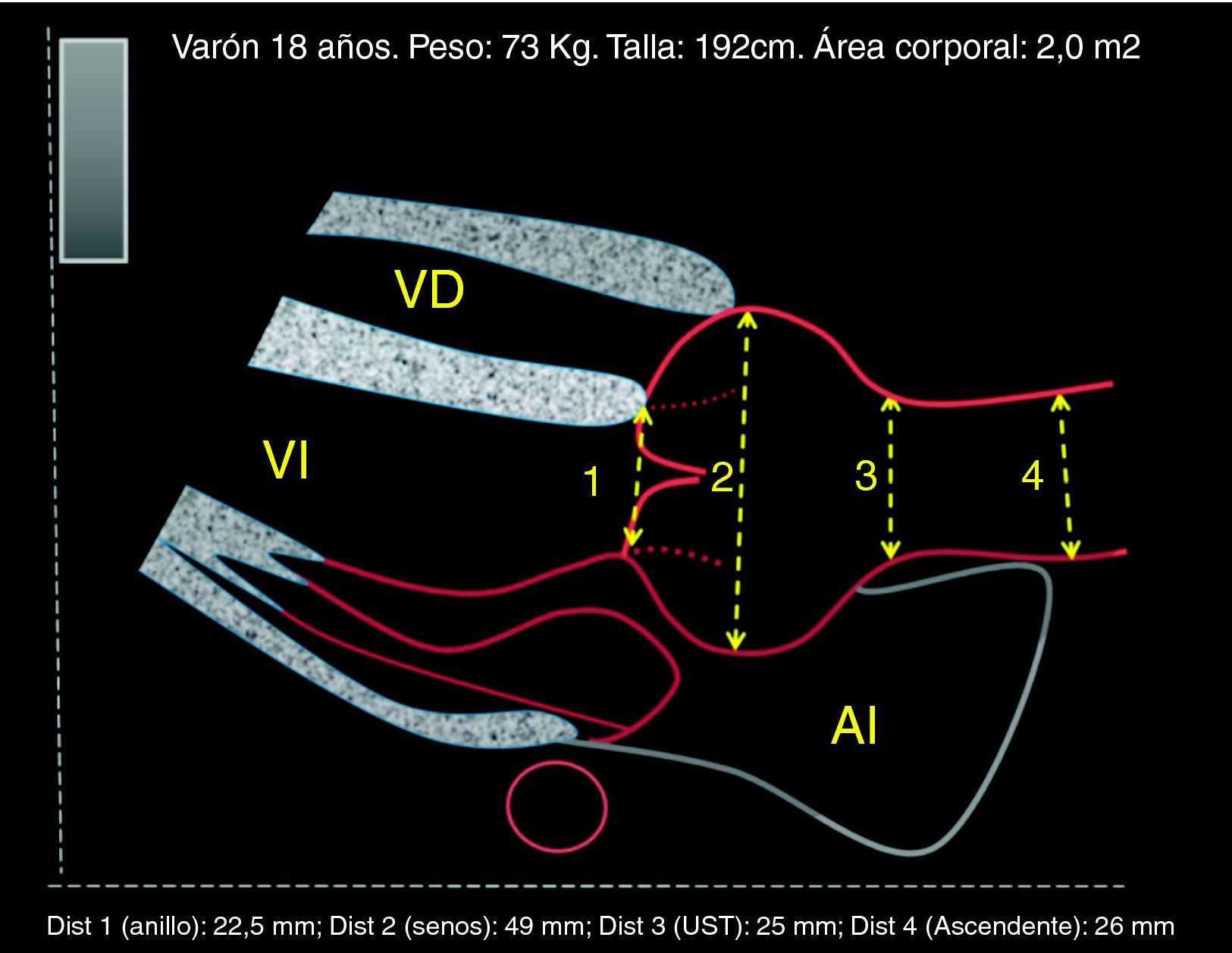
Respecto al timing de la cirugía electiva, se deben hacer algunas consideraciones. Según el valor del umbral de diámetro, una proporción más o menos importante de pacientes presentarán complicaciones sin alcanzar este valor o se someterán injustificadamente al riesgo quirúrgico de un procedimiento electivo estando alejados aún de complicaciones. Resulta importante incorporar otra información, como el ritmo de crecimiento, y ajustar diámetros por el tamaño corporal. La SC, empleada en muchos nomogramas al contemplar el peso, puede modificar artificialmente el umbral quirúrgico. La tendencia actual es corregir según la estatura, para que en sujetos de menor estatura —especialmente mujeres— pero con riesgo de complicación pueda indicarse cirugía aun cuando sus diámetros estén más próximos a 45 que a 50 mm (fig. 4). En la práctica, la indicación quirúrgica se empieza a considerar cuando la aorta está dilatada (≥2 desviaciones por encima de la media, Z-score ≥2) o cuando se aproxima a 45 mm de diámetro (antes si la estatura es inferior a 170 cm). Los resultados quirúrgicos son determinantes para indicar cirugía profiláctica, preferentemente conservando la válvula y con muy baja mortalidad, obligatoriamente inferior al 5%7,8.
. Acude a control en el quinto mes de gestación, la paciente no se quería operar antes de decidir el embarazo. La raíz aórtica con <40mm, ratio 1,3, no haría recomendable la cirugía electiva, según las guías. A los 3 meses postparto la raíz ha crecido hasta 45mm, la ratio de 1,5 y la tasa de crecimiento aórtico harían recomendable la intervención electiva sobre la raíz. No obstante, debido a la ganancia de peso, el Z-score habría disminuido. LSN: límite superior de la normalidad.")
Toma de decisiones sobre la cirugía electiva. La toma de decisiones puede ser muy compleja, a veces con contradicciones entre criterios, como se puede ver en este ejemplo hipotético: Mujer >18 años con SM y gestación avanzada (no siguió recomendaciones). Acude a control en el quinto mes de gestación, la paciente no se quería operar antes de decidir el embarazo. La raíz aórtica con <40mm, ratio 1,3, no haría recomendable la cirugía electiva, según las guías. A los 3 meses postparto la raíz ha crecido hasta 45mm, la ratio de 1,5 y la tasa de crecimiento aórtico harían recomendable la intervención electiva sobre la raíz. No obstante, debido a la ganancia de peso, el Z-score habría disminuido. LSN: límite superior de la normalidad.
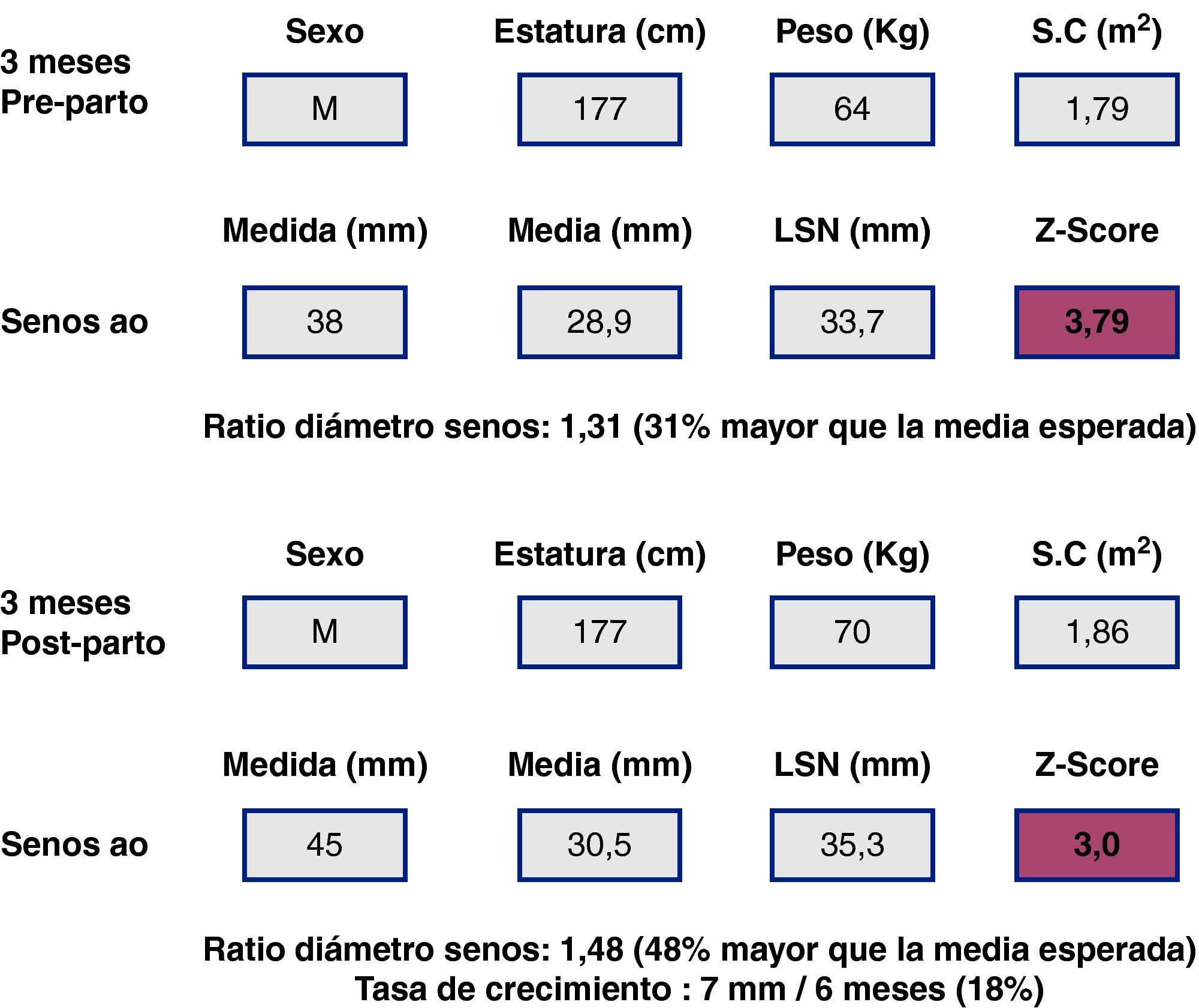
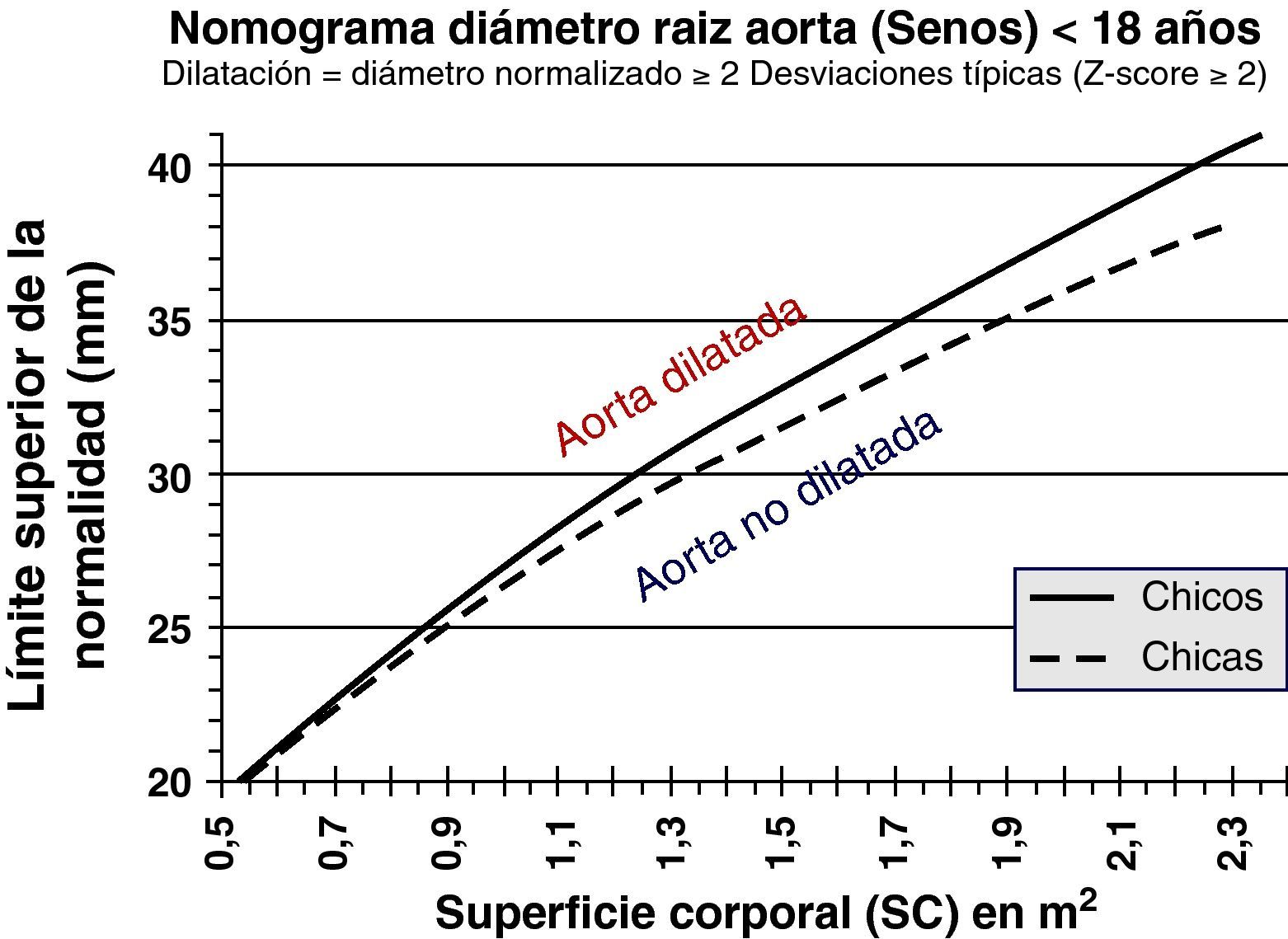
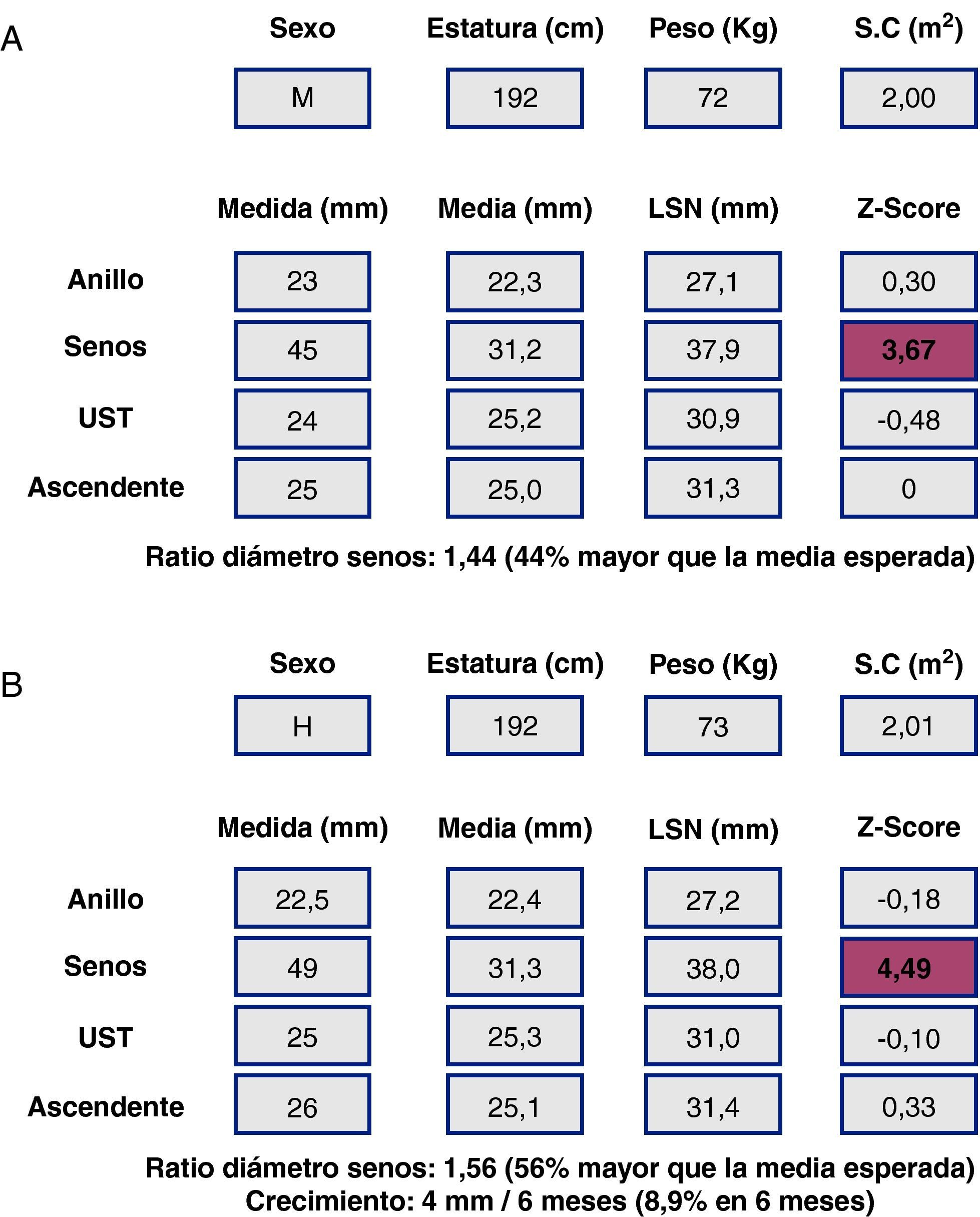
Al ser las complicaciones aórticas infrecuentes antes de los 12 años, establecer una relación con el diámetro es más difícil que en adultos. La cirugía electiva de la aorta en población infantil (hasta 18 años) con SM se recomienda cuando el diámetro aórtico excede los 50 mm, ante el rápido crecimiento aórtico (>10mm/año), ante el desarrollo de IAo, o cuando hay afectación simultánea de la válvula mitral. En cuanto al timing, es necesario sopesar el riesgo de disección y el retraso del momento quirúrgico para evitar mismatch protésico, ya que los niños seguirán creciendo. Los nomogramas pediátricos han sido recalculados para mejorar su correspondencia con los de adultos (fig. 5). La normalización por sexo, edad y SC parece adecuada, aunque habrá que definir mejor cuál es la dilatación de riesgo en la que los beneficios de la cirugía profiláctica inequívocamente superan los riesgos. En la figura 6 se presentan ejemplos hipotéticos en la toma de decisiones.

 y cirugía electiva de la raíz aórtica: toma de decisiones y timing. Caso hipotético. Varón de 18 años. Panel superior (A), SM, datos de medidas aórticas en el primer estudio ecocardiográfico y en el estudio a los 6 meses (panel inferior, B). La poca adiposidad que caracteriza a los pacientes más jóvenes con SM hace que, para una misma estatura y diámetro aórtico, el Z-score y la ratio en porcentaje sean mayores cuanto más delgado se está. De ahí que algunos expertos opinen que es preferible en el SM corregir los diámetros tan sólo teniendo en cuenta la estatura.")
Síndrome de Marfan (SM) y cirugía electiva de la raíz aórtica: toma de decisiones y timing. Caso hipotético. Varón de 18 años. Panel superior (A), SM, datos de medidas aórticas en el primer estudio ecocardiográfico y en el estudio a los 6 meses (panel inferior, B). La poca adiposidad que caracteriza a los pacientes más jóvenes con SM hace que, para una misma estatura y diámetro aórtico, el Z-score y la ratio en porcentaje sean mayores cuanto más delgado se está. De ahí que algunos expertos opinen que es preferible en el SM corregir los diámetros tan sólo teniendo en cuenta la estatura.
Aunque la cirugía electiva de la aorta descendente es actualmente más segura, continúa existiendo riesgo de paraplejía (que debería ser inferior al 5%) dependiente de la experiencia del grupo, de la extensión del segmento aórtico a reemplazar y de la protección medular. Puesto que el riesgo operatorio aumenta en la emergencia (disección o rotura), y dada la limitación para el uso de stents en estos pacientes, se recomienda el reemplazo profiláctico del segmento aórtico comprometido cuando el diámetro es >55 mm (recomendación clase I, nivel de evidencia: C)1,7.
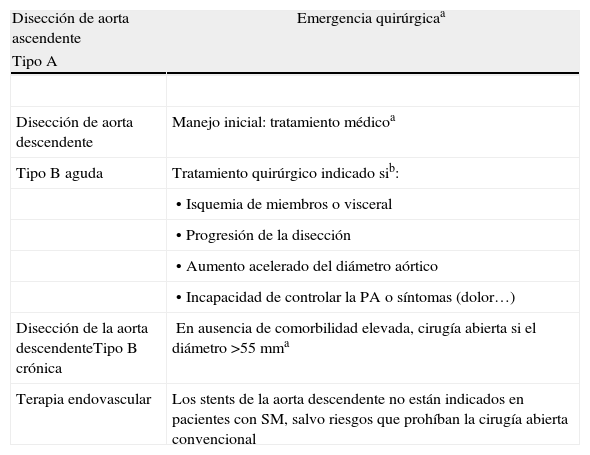
Tratamiento de las complicaciones aórticas agudas (tabla 6)Disección de aorta ascendente (tipo A)Dada la naturaleza impredecible de la disección aórtica en el SM, hay que educar a los pacientes sobre los síntomas de la disección aguda. Como en la población general, la disección aguda tipo A en el SM es una emergencia quirúrgica en la que deben reemplazarse los senos y la suficiente extensión de la aorta ascendente.
Tratamiento de complicaciones aórticas en el síndrome de Marfan
| Disección de aorta ascendente | Emergencia quirúrgicaa |
| Tipo A | |
| Disección de aorta descendente | Manejo inicial: tratamiento médicoa |
| Tipo B aguda | Tratamiento quirúrgico indicado sib: |
| • Isquemia de miembros o visceral | |
| • Progresión de la disección | |
| • Aumento acelerado del diámetro aórtico | |
| • Incapacidad de controlar la PA o síntomas (dolor…) | |
| Disección de la aorta descendenteTipo B crónica | En ausencia de comorbilidad elevada, cirugía abierta si el diámetro >55 mma |
| Terapia endovascular | Los stents de la aorta descendente no están indicados en pacientes con SM, salvo riesgos que prohíban la cirugía abierta convencional |
aRecomendación clase I, nivel de evidencia B.
bManejo posterior: betabloqueo, medicación adicional si es necesaria para el control de la PA, y seguimiento con RM o TC según los síntomas, el diámetro y el ritmo de crecimiento aórtico.
La disección tipo B representa aproximadamente un 10% de las disecciones agudas en el SM. Como en otros pacientes, inicialmente se recomienda el tratamiento médico, salvo complicación o falta de respuesta, en cuyo caso debe considerarse la cirugía. Se recomienda la realización rutinaria de TC o angio-RM de la aorta completa si la descendente es grande o si se ha disecado tras la reparación de una disección tipo A. En la disección crónica tipo B se recomienda la cirugía abierta cuando, en ausencia de comorbilidad importante, se superan los 55 mm.
Terapia endovascular: stentsAunque la experiencia con endoprótesis en la disección aórtica tipo B aguda o crónica en el SM es limitada, se ha observado que a pesar del correcto implante del stent, con trombosis total de la falsa luz, la aorta continúa dilatándose. Por este motivo se recomienda no utilizar stents aórticos en el SM, salvo riesgo prohibitivo para la cirugía convencional. Los seudoaneurismas tras reemplazo aórtico pueden ser una excepción cuando es posible anclar al injerto previo un stent que selle el «cuello» del falso aneurisma como alternativa a la re-toracotomía.
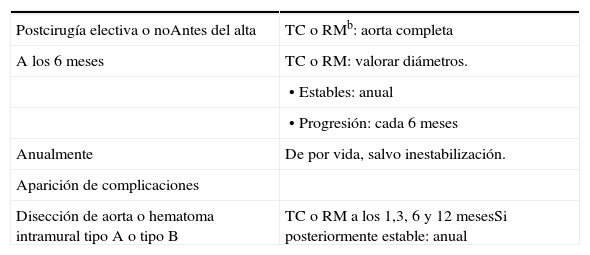
Recomendaciones tras intervención aórtica en el SMTras la reparación de la aorta, los injertos, relativamente rígidos, transmiten tensión hacia territorios contiguos: coronarias, cayado y troncos principales y la aorta descendente, predisponiendo al desarrollo tardío de aneurismas y disección. Los pacientes deben mantener los betabloqueantes y ser seguidos mediante técnicas de imagen de por vida, restringiendo la irradiación por TC cuando sea posible (tabla 7).
Seguimiento tras cirugía aórtica en el síndrome de Marfana
| Postcirugía electiva o noAntes del alta | TC o RMb: aorta completa |
| A los 6 meses | TC o RM: valorar diámetros. |
| • Estables: anual | |
| • Progresión: cada 6 meses | |
| Anualmente | De por vida, salvo inestabilización. |
| Aparición de complicaciones | |
| Disección de aorta o hematoma intramural tipo A o tipo B | TC o RM a los 1,3, 6 y 12 mesesSi posteriormente estable: anual |
aRecomendaciones clase IIa, nivel de evidencia C.
bLa aorta debe ser valorada en su totalidad, no solamente la porción ascendente, ya que una gran proporción (casi un tercio) de los eventos aórticos que comprometen la aorta distal ocurren durante el seguimiento de estos pacientes.
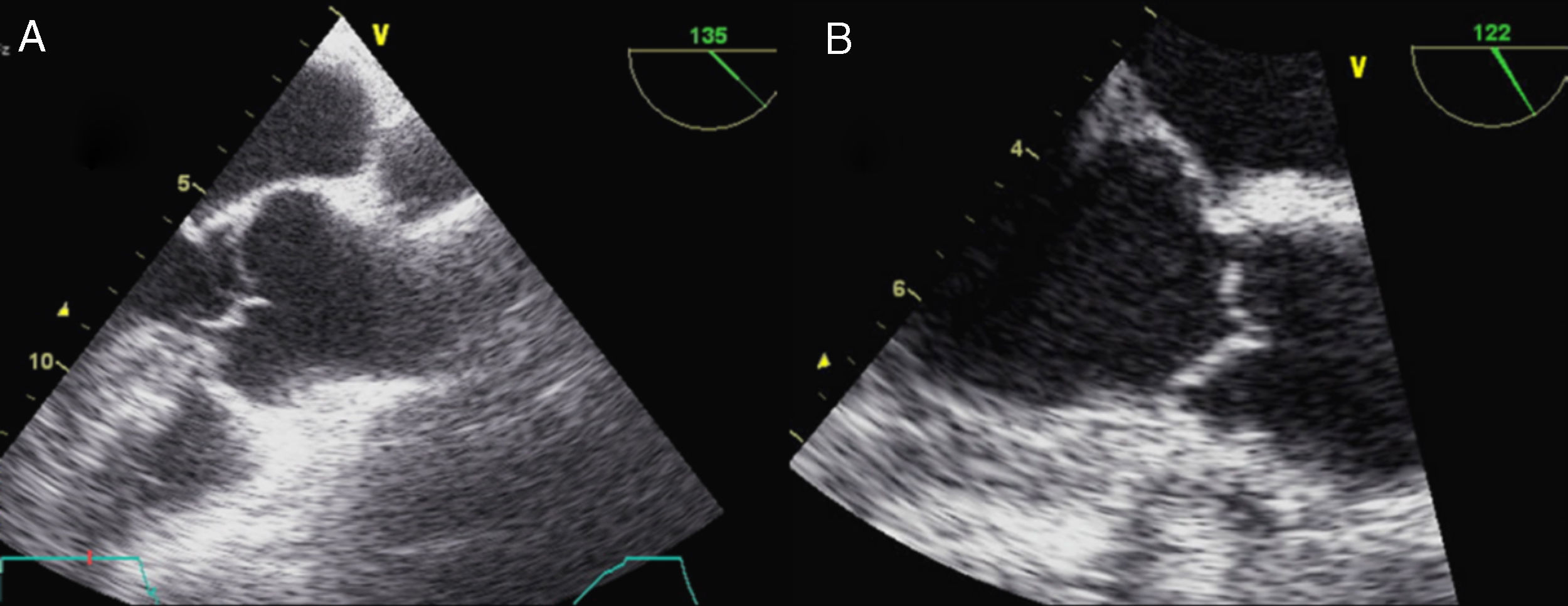
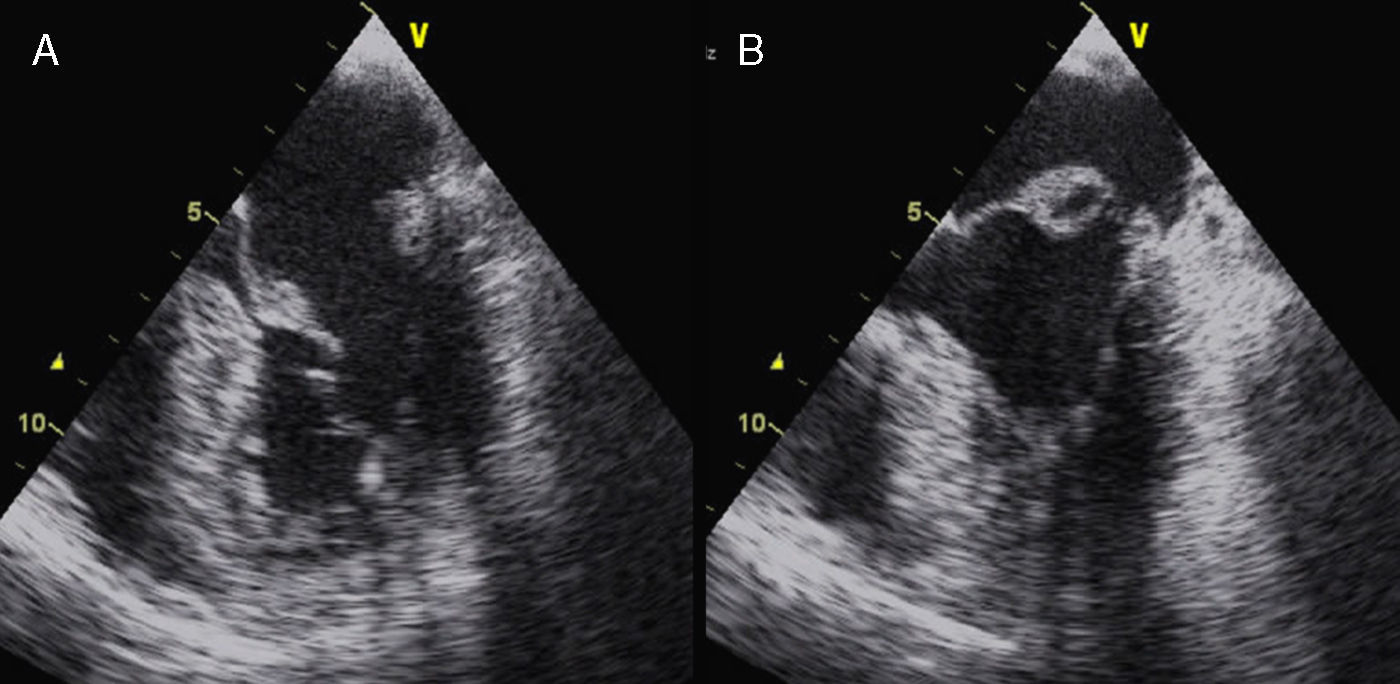
La afectación mitral y la tricúspide constituyen el hallazgo cardiaco más frecuente en el SM, aunque la tricúspide raramente tiene repercusión. Las alteraciones del tejido conectivo mitral conducen al crecimiento con aspecto mixoide, con elevado contenido aéreo en su interior, si bien la histología y la morfología de la mitral en pacientes con SM son diferentes de la afectación valvular mixoide clásica. En el SM los velos, aunque más gruesos de lo normal (fig. 7), son más largos y delgados que las mixoides y con menor celularidad2. Los pacientes con SM presentan más frecuentemente compromiso de ambos velos o del velo anterior, lo que, junto con la laxitud del aparato valvular, hace que el prolapso mitral sea más frecuente en el SM. En estos pacientes el prolapso puede producir insuficiencia mitral moderada o mayor hasta en el 25% de los casos. También es característica la tendencia a la calcificación temprana del anillo, lo que constituye un criterio diagnóstico menor. En las formas más graves del SM, que se inician en los primeros años de vida, la afectación mitral puede causar insuficiencia cardíaca e hipertensión pulmonar con resultados quirúrgicos muy desfavorables en menores de 2 años, siendo una causa de mortalidad importante en niños con SM. En adolescentes y adultos la reparación quirúrgica de la insuficiencia mitral severa se asocia con una elevada supervivencia libre de eventos. La cirugía mitral aislada es infrecuente, y la mayoría de las ocasiones se realizan procedimientos combinados reparadores sobre la aorta y la mitral para evitar la anticoagulación. La calcificación extensa del anillo mitral es la principal contraindicación para la reparación mitral en el SM. Es importante insistir que la insuficiencia mitral severa no corregida afecta adversamente al estrés hemodinámico aórtico y a la función ventricular en el SM.
. A) En telediástole se aprecia el aumento del grosor valvular con elongación de cuerdas. B) En telesístole se observa abombamiento de ambos velos, con prolapso amplio de la valva anterior.")
Afectación mitral en el síndrome de Marfan. Ecocardiograma transesofágico, proyección del eje largo de 4 cámaras (0°). A) En telediástole se aprecia el aumento del grosor valvular con elongación de cuerdas. B) En telesístole se observa abombamiento de ambos velos, con prolapso amplio de la valva anterior.
La dilatación de la raíz del tronco de la arteria pulmonar es menos frecuente que la aórtica, y raramente provoca disección. En el SM podrían darse alteraciones de la conducción auriculoventricular y de la repolarización ventricular (QT prolongado, alteración del ST y ondas U), que podrían asociarse a arritmias ventriculares, pero no está claro si estas alteraciones son secundarias a una miocardiopatía primaria genuina o a dilatación ventricular debida a regurgitaciones evolucionadas5.
ConclusionesEn las últimas décadas se han producido importantes modificaciones en el pronóstico del SM, que además del riesgo de muerte súbita ocasiona múltiples trastornos. El manejo cardiovascular de estos pacientes se basa en tres pilares dirigidos a aumentar la esperanza y la calidad de vida: estratificación del riesgo, tratamiento médico y cirugía aórtica profiláctica.
Las técnicas de imagen contribuyen a estratificar el riesgo y a seleccionar mejor los casos y el momento más adecuados para la cirugía electiva.
Todos los pacientes deben ser tratados tempranamente, al menos, con betabloqueantes. Mientras tanto, seguirán evaluándose nuevas terapias, dirigidas no a retrasar las complicaciones sino a detener o incluso revertir cambios patológicos.
Cada vez más pacientes con SM llegarán a etapas avanzadas de la vida y plantearán nuevos retos: pacientes intervenidos, enfermedad mitral más evolucionada, patología de la aorta descendente… Se pondrá a prueba el conocimiento adquirido, y será imprescindible el trabajo en equipo desde unidades multidisciplinares especializadas. La atención integral a pacientes con SM, como ya hemos aprendido, es algo más que disponer de un programa de cirugía de la aorta.

