





El síndrome de Marfan (SMF) es una enfermedad del tejido conectivo con herencia autosómica dominante que afecta principalmente al sistema cardiovascular, ocular y musculoesquelético; se caracteriza por una alta penetrancia y su marcada heterogeneidad fenotípica.
La prevalencia estimada es de 1 por cada 5.000-10.000 nacidos vivos, con afectación similar por sexo. En la mayoría de los casos el SMF está causado por una mutación en el gen de la fibrilina-1 (FBN1), situado en el cromosoma 15 (15q21.1)1, glucoproteína ampliamente distribuida en tejidos elásticos y no elásticos. A pesar de los avances en el conocimiento acerca de la genética en el SMF, los mecanismos moleculares que dan lugar al desarrollo del fenotipo no están claramente dilucidados.
Descrito por primera vez en 1896 por el pediatra francés Antoine-Bernard Marfan2 e incluido en 1955 en una clasificación de enfermedades del tejido conectivo3, no fue hasta 1986 cuando un panel internacional de expertos4 definió un conjunto de criterios clínicos (nosología de Berlín) para el diagnóstico del SMF, con una modificación posterior en 19965, referida desde entonces como nosología de Gante (Ghent nosology). Esta última, que incluyó la presencia de mutaciones en el gen FBN1, y con unos criterios más restrictivos que los de la nosología de Berlín, tuvo como objetivos disminuir el sobrediagnóstico del síndrome y facilitar mejores guías para diferenciarlo de otras entidades que se superponían.
Los criterios Gante (tabla 1) han sido mundialmente utilizados, ayudando a los profesionales en el diagnóstico del SMF, con una altísima especificidad, al haberse detectado mutaciones en FBN1 hasta en el 97% de los pacientes que reúnen estos criterios6. Sin embargo, presenta también limitaciones, tales como el hecho de no tener suficientemente en cuenta la dependencia de la edad en algunas manifestaciones clínicas (haciendo difícil el diagnóstico en niños), o incluir algunas manifestaciones físicas no específicas o con valor diagnóstico escasamente validado. Esto puede dar lugar a diagnosticar erróneamente de SMF a pacientes con síndrome de ectopia lentis (SEL), síndrome de prolapso de válvula mitral (SPVM) o el fenotipo MASS, o por el contrario, no hacerlo en pacientes con ectopia lentis y dilatación aórtica sin suficientes manifestaciones esqueléticas.
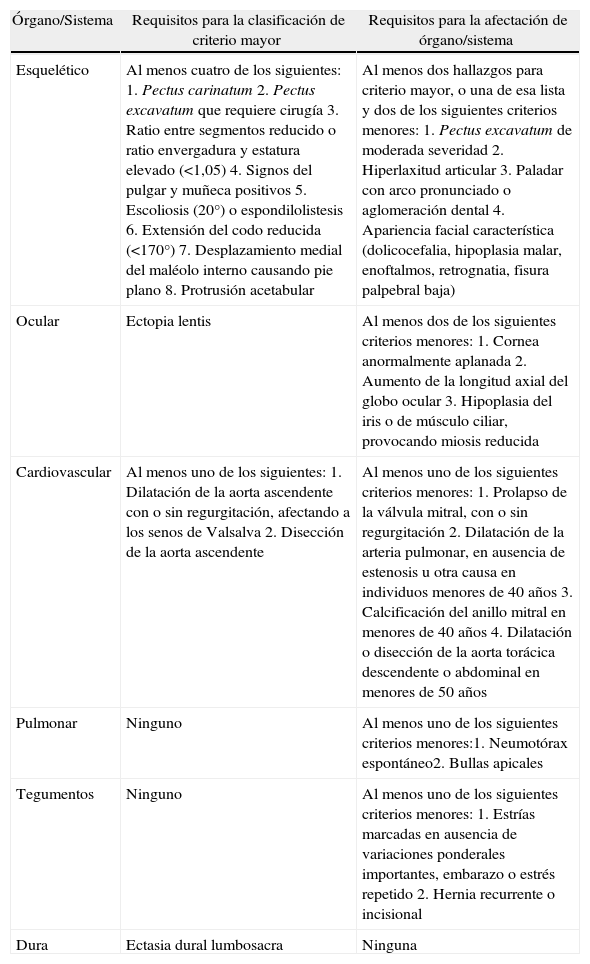
Criterios diagnósticos de la nosología de Gante
| Órgano/Sistema | Requisitos para la clasificación de criterio mayor | Requisitos para la afectación de órgano/sistema |
| Esquelético | Al menos cuatro de los siguientes:1. Pectus carinatum2. Pectus excavatum que requiere cirugía3. Ratio entre segmentos reducido o ratio envergadura y estatura elevado (<1,05)4. Signos del pulgar y muñeca positivos5. Escoliosis (20°) o espondilolistesis6. Extensión del codo reducida (<170°)7. Desplazamiento medial del maléolo interno causando pie plano8. Protrusión acetabular | Al menos dos hallazgos para criterio mayor, o una de esa lista y dos de los siguientes criterios menores:1. Pectus excavatum de moderada severidad2. Hiperlaxitud articular3. Paladar con arco pronunciado o aglomeración dental4. Apariencia facial característica (dolicocefalia, hipoplasia malar, enoftalmos, retrognatia, fisura palpebral baja) |
| Ocular | Ectopia lentis | Al menos dos de los siguientes criterios menores:1. Cornea anormalmente aplanada2. Aumento de la longitud axial del globo ocular3. Hipoplasia del iris o de músculo ciliar, provocando miosis reducida |
| Cardiovascular | Al menos uno de los siguientes:1. Dilatación de la aorta ascendente con o sin regurgitación, afectando a los senos de Valsalva2. Disección de la aorta ascendente | Al menos uno de los siguientes criterios menores:1. Prolapso de la válvula mitral, con o sin regurgitación2. Dilatación de la arteria pulmonar, en ausencia de estenosis u otra causa en individuos menores de 40 años3. Calcificación del anillo mitral en menores de 40 años4. Dilatación o disección de la aorta torácica descendente o abdominal en menores de 50 años |
| Pulmonar | Ninguno | Al menos uno de los siguientes criterios menores:1. Neumotórax espontáneo2. Bullas apicales |
| Tegumentos | Ninguno | Al menos uno de los siguientes criterios menores:1. Estrías marcadas en ausencia de variaciones ponderales importantes, embarazo o estrés repetido2. Hernia recurrente o incisional |
| Dura | Ectasia dural lumbosacra | Ninguna |
Para el diagnóstico de síndrome de Marfan en pacientes sin antecedentes familiares de enfermedad deben estar involucrados dos órganos/sistemas que reúnan criterios mayores y al menos la afectación de un tercer órgano/sistema. En pacientes con historia familiar de síndrome de Marfan solo se requiere un criterio mayor, con datos que sugieran afectación de un segundo sistema. (De Paepe et al5.)
Con objeto de salvar las limitaciones que se han detectado en la nosología de Gante, así como por la necesidad de definir mejor ciertas categorías diagnósticas, identificar individuos que podrían tener diagnóstico alternativo o concretar guías de manejo para varios grupos de pacientes, tales como niños que no reúnen criterios suficientes pero que pueden hacerlo en el futuro, se ha propuesto una revisión de la nosología Gante.
Un panel internacional de expertos en el diagnóstico y manejo del SMF convocado en Bruselas por la National Marfan Foundation (EE.UU.) propuso y publicó recientemente la nosología Gante revisada (the revised Ghent nosology)7 basándose en la revisión crítica de amplias cohortes de pacientes publicadas y las opiniones expertas de los miembros del panel, con extensa experiencia en la aplicación de los criterios clásicos, en el diagnóstico diferencial del SMF, y en la solidez y las limitaciones del estudio genético.
Cambios mayores en las guías diagnósticas8- •
Se concede mayor valor a dos hallazgos cardinales del SMF, el aneurisma/disección de la raíz aórtica y la ectopia lentis, siendo suficiente la combinación de ambas para establecer el diagnóstico. El resto de manifestaciones oculares y cardiovasculares, así como los hallazgos de otros órganos/sistemas, contribuyen a un score sistémico (tabla 2) que guía el diagnóstico cuando está presente la enfermedad aórtica pero no la ectopia lentis.
Tabla 2.Score de hallazgos sistémicos
Signo de la muñeca y el pulgar: 3 (signo de la muñeca o pulgar: 1) Pectus carinatum: 2 (pectus excavatum o asimetría pectoral: 1) Deformidad retropié: 2 (pie plano: 1) Neumotórax: 2 Ectasia dural: 2 Protrusión acetabular: 2 SS/SI reducida y ratio brazo/estatura incrementada y escoliosis no severa: 1 Escoliosis o cifosis toracolumbar: 1 Extensión reducida del codo: 1 Hallazgos faciales (3/5): 1 (dolicocefalia, enoftalmos, fisura palpebral baja, hipoplasia malar, retrognatia) Estría cutánea: 1 Miopía >3 dioptrías: 1 Prolapso mitral (todos los tipos): 1 Total máximo 20 puntos; un score ≥7 indica afectación sistémica.
MS/MI: ratio segmento superior/inferior.
- •
Al estudio genético molecular de FBN1 y otros genes relevantes (p. ej., TGFBR1 y TGFBR2) se le asigna un papel más prominente. En la práctica no es requisito formal (teniendo en cuenta la carga económica que impone y la ausencia de un 100% de sensibilidad y especificidad), pero se considera adecuado cuando esté disponible.
- •
Algunas de las manifestaciones del SMF menos específicas pierden importancia en la evaluación diagnóstica.
- •
Los nuevos criterios formalizan el concepto de requerirse consideraciones diagnósticas y pruebas adicionales si los pacientes reúnen suficientes criterios para SMF pero muestran hallazgos inesperados, sobre todo ante la posibilidad de un diagnóstico alternativo específico. Se enfatiza especialmente en los síndromes de Sphrintzen-Goldberg (SSG) y de Loeys-Dietz (SLD), y en la forma vascular del síndrome de Ehlers-Danlos (SEDv).
En la nosología revisada, los nuevos criterios diagnósticos se han definido para un paciente índice esporádico o para un paciente con una historia familiar positiva (tabla 3).
- A.
En ausencia de historia familiar de SMF, el diagnóstico puede ser establecido en cuatro escenarios distintos:
- 1.
La presencia de disección o dilatación de la raíz aórtica (Z-score ≥2, ajustado a edad y superficie corporal)8 y ectopia lentis indica el diagnóstico de SMF, independientemente de la existencia de hallazgos sistémicos, salvo cuando éstos sean indicativos de SSG, SLD o SEDv.
- 2.
La presencia de disección o dilatación (Z-score ≥2) y la identificación de una mutación causal en FBN1 es suficiente para establecer el diagnóstico, aun en ausencia de ectopia lentis.
- 3.
En presencia de disección o dilatación (Z-score ≥2), sin ectopia lentis y ausencia o desconocimiento de mutaciones en FBN1, puede establecerse el diagnóstico de SMF cuando existan suficientes hallazgos sistémicos (≥7 puntos), aunque se deben excluir en este caso la posibilidad de SSG, SLD o SEDv con los estudios correspondientes.
- 4.
En presencia de ectopia lentis pero sin dilatación/disección aórtica, la identificación de una mutación en FBN1 previamente asociada a enfermedad aórtica permite confirmar el SMF. Si la mutación en FBN1 no está asociada a enfermedad cardiovascular el paciente sería clasificado como síndrome de ectopia lentis.
- 1.
- B.
En caso de individuos con historia familiar de SMF, el diagnóstico puede ser establecido por la presencia de ectopia lentis, un score sistémico ≥7 puntos o la presencia de dilatación de la raíz aórtica (Z ≥2 en adultos ≥20 años, o Z ≥3 en individuos <20 años).
Criterios Ghent revisados para diagnóstico de síndrome de Marfan
| En ausencia de historia familiar de síndrome de Marfan |
| 1. Ao (Z ≥2) y EL=SMFa |
| 2. Ao (Z ≥2) y mutación FBN1=SMF |
| 3. Ao (Z ≥2) y score sistémico (≥7 puntos)=SMFa |
| 4. EL y FBN1 identificada en individuos con aneurisma aórtico=SMF |
| • EL con o sin score sistémico, sin mutación en FBN1, o con mutación FBN1 no relacionada con aneurisma/disección aórtica=SEL |
| • Ao (Z ≥2) y score sistémico (Z ≥5) sin EL=MASS |
| • PVM y Ao (Z <2) y score sistémico (<5) sin EL=SPVM |
| En presencia de historia familiar (HF) de síndrome de Marfan |
| 5. EL y HF de SMF=SMF |
| 6. Score sistémico ≥7 puntos y HF de SMF=SMFa |
| 7. Ao (Z ≥2 en mayores de 20 años, Z ≥3 en menores de 20 años) e HF de SMF=SMFa |
Ao: diámetro aórtico en senos de Valsalva (indicado por Z-score) o disección; mutación FBN1: mutación en fibrilina 1; EL: ectopia lentis; MASS: fenotipo con miopía, prolapso mitral, dilatación limítrofe de raíz aórtica (Z<2), estrías y hallazgos esqueléticos; PVM: prolapso de válvula mitral; SEL: síndrome de ectopia lentis; SMF: indica síndrome de Marfan; SPVM: síndrome de prolapso de válvula mitral; Z: Z-score.
Además, se consideran dos nuevas situaciones en menores de 20 años. En primer lugar, el «trastorno inespecífico del tejido conectivo» para los casos con insuficientes hallazgos sistémicos (<7) y/o dimensiones limítrofes de la raíz aórtica (Z <3), sin mutación de FBN1. En segundo lugar, el término «SMF potencial» se aplicaría en los casos, esporádicos o familiares, con mutación identificada en FBN1 pero dimensiones que no alcanzan un Z score de 3.
En adultos, se definen además tres categorías alternativas: síndrome de ectopia lentis (SEL), síndrome de prolapso de válvula mitral (SPVM) y el fenotipo MASS.
Finalmente, reconocen que algunos pacientes son difíciles de clasificar debido a la superposición de fenotipos en distintas entidades.
Consideraciones específicasCriterios cardiovascularesUn criterio diagnóstico clave en la nueva nosología es la disección o dilatación de la raíz aórtica. El aneurisma es definido como la dilatación de la raíz aórtica al nivel de los senos de Valsalva. La mayor medida de la raíz obtenida correctamente debe ser corregida según la edad y la superficie corporal e interpretada como un Z-score8. Si la evaluación ecocardiográfica transtorácica no permite una adecuada visualización de la aorta proximal, se deben aplicar otras técnicas de imagen, tales como el ecocardiograma transesofágico, la tomografía computarizada o la resonancia magnética y aplicar los nomogramas correspondientes.
El prolapso de la válvula mitral se incluye en el score sistémico, sin criterio específico en su diagnóstico, debiéndose aplicar la práctica habitual.
Aunque la dilatación o disección de la aorta torácica en ausencia de dilatación de la raíz puede suceder en el SMF, esta es excepcional y, dada la baja especificidad, no se incluye entre los criterios diagnósticos. Sin embargo, el estudio de imagen intermitente en la aorta torácica descendente está indicado en individuos con sospecha de SMF, aun en ausencia de dilatación de la raíz.
Criterios ocularesLos hallazgos oculares fundamentales en el SMF son la miopía y la ectopia lentis. Esta última se basa en la exploración con lámpara de hendidura bajo dilatación máxima de la pupila. La miopía es muy frecuente y suele ser de presentación precoz y rápidamente progresiva, de modo que un defecto superior a 3 dioptrías contribuye al diagnóstico en el score sistémico, si bien dado que es un hallazgo habitual en la población general solo se le atribuye un punto es tal score.
Criterios sistémicosLas manifestaciones clínicas del SMF en otros órganos y sistemas fueron críticamente evaluadas por su especificidad y utilidad diagnóstica basada en la opinión experta y la literatura disponible. Varios de los criterios «menores» de la nosología de Ghent se eliminan, pero los hallazgos sistémicos más selectivos se incluyen en el score sistémico (tabla 2).
A la combinación de los signos de la muñeca y el pulgar se les asigna tres puntos. El signo del pulgar es positivo cuando la falange distal del mismo se extiende al borde cubital de la palma, con o sin ayuda del paciente o examinador para lograr la máxima abducción. El signo de la muñeca es positivo cuando la punta del pulgar cubre enteramente las uñas de los cinco dedos cuando envuelve la muñeca contralateral. Si uno de los dos signos está ausente, solo se asigna un punto.
Se asignan dos puntos a la deformidad anterior del tórax, deformidad del retropié, neumotórax espontáneo, ectasia dural y protrusión acetabular. El pectus carinatum se considera más específico del SMF que el pectus excavatum, asignándosele 2 puntos.
Se asigna un punto a ocho manifestaciones, que incluyen una cardiovascular (prolapso de válvula mitral), una ocular (miopía ≥3 dioptrías) y seis de otros órganos/sistemas. Estas son menos específicas y pueden observarse en otros trastornos del tejido conectivo o como variación normal en la población general.
La presencia de una relación reducida entre segmento superior y segmento inferior (<0,85 para adultos blancos) y ratio envergadura y estatura incrementada (>1,05 para adultos) contribuye en un solo punto en el score sistémico. La escoliosis de al menos 20° de ángulo de Cobb o una exagerada cifosis toracolumbar, una extensión reducida del codo, los hallazgos faciales (dolicocefalia, fisura palpebral baja, enoftalmos, retrognatia e hipoplasia malar) y la presencia de estrías atróficas (no asociadas con cambios de peso acentuados) tienen asignados un punto cada una.
Se eliminan así de la nosología actual la hiperlaxitud articular, el arco paladar aumentado y la hernia recurrente o incisional.
Criterio genéticoEn la nosología revisada se otorga un peso mayor a la genética en el diagnóstico de SMF y los síndromes relacionados, estableciéndose criterios de causalidad a las mutaciones en FBN1. En el caso de que la mutación haya sido descrita previamente, debe demostrarse cosegregación familiar. Si la mutación no está descrita previamente, se deben tener en cuenta que tenga probabilidad de ser patogénica (mutación sin sentido o nonsense, inserción/deleción, mutaciones splice site o missense de determinadas características, etc.), y debe demostrarse, si es posible, cosegregación en la familia.
Diagnóstico diferencialHay varias entidades en las que se reconocen manifestaciones clínicas que se superponen con el SMF en los sistemas cardiovascular, ocular y esquelético. Esto incluye entidades con aneurisma aórtico (SLD, válvula aórtica bicúspide, aneurisma aórtico torácico familiar, SEDv, síndrome de tortuosidad arterial), ectopia lentis (síndrome de ectopia lentis, síndrome de Weil-Marchesani, homocistinuria y síndrome de Stickler) o manifestaciones sistémicas del SMF (síndrome de Shprintzen-Goldberg, aracnodactilia contractural congénita o síndrome de Beals, SLD, fenotipo MASS y SPVM).
ConclusionesLa evaluación diagnóstica del SMF es inevitablemente compleja debido a la alta variabilidad de presentación de los individuos afectados, la dependencia de la edad en muchas de las manifestaciones, ausencia de gold standards, y el amplio diagnóstico diferencial. Esta nueva propuesta en el diagnóstico del SMF pretende facilitar una correcta y precoz identificación por los profesionales que la atienden y así mejorar el pronóstico de los pacientes.

