



Las nuevas técnicas desarrolladas para el análisis genético nos aportan información sobre las bases genéticas de las distintas miocardiopatías, entre ellas, de la miocardiopatía dilatada familiar. Realizamos esta revisión con el fin de aportar información sobre la utilidad clínica del estudio genético en el manejo de estos pacientes.
The new technologies developed for genetic analysis provide information on the genetics basis of the different types of cardiomyopathy, including familial dilated cardiomyopathy. A review is presented with the aim of providing information on the clinical use of the genetic study in the management of these patients.
Las miocardiopatías son aquellas afecciones cardíacas que cursan con una alteración estructural y funcional del miocardio en ausencia de otras enfermedades que las justifiquen (cardiopatía congénita, cardiopatía isquémica, valvulopatía o hipertensión arterial). La actual clasificación se atiene a sus distintas características morfológicas para subclasificarlas posteriormente en función de que exista o no agregación familiar o causa genética. Dentro de esta clasificación se define la miocardiopatía dilatada (MCD) según WHO/International Society and Federation of Cardiology Task Force como «la dilatación de la cavidad ventricular que supone una afectación sistólica en ausencia de otras condiciones anormales o enfermedad arterial coronaria suficiente para causar disfunción sistólica global»1.
La MCD es una importante causa de insuficiencia cardíaca, la causa más frecuente de trasplante cardíaco y, según series recientes, la MCD familiar supone entre el 30 y el 50% de todas las MCD2. En los últimos tiempos los avances en genética están arrojando luz sobre las bases moleculares de esta y otras enfermedades cardíacas, y paulatina e irreversiblemente estos conocimientos se están trasladando a la clínica diaria, aún con las incertidumbres inherentes al manejo práctico de esta nueva herramienta. La MCD familiar es generalmente una enfermedad monogénica, aunque existe en ella una gran heterogeneidad genética, así como un importante solapamiento con el espectro genético del resto de miocardiopatías, por lo que se están haciendo importantes esfuerzos en la comprensión de sus bases genéticas. Según algunos autores, el número de genes implicados sería mayor de 303, aunque otros autores elevan esta cifra por encima de los 404. Estos genes codifican proteínas con diversas funciones estructurales dentro del músculo cardíaco, distinguiéndose así entre proteínas sarcoméricas, desmosómicas, nucleares, del citoesqueleto, de la membrana y de los canales iónicos. Las alteraciones en estas proteínas conllevan cambios estructurales y funcionales a nivel del miocito que afectan a la fuerza contráctil en el sarcómero, la transmisión de la fuerza contráctil o la integridad celular4.
La llegada de la ultrasecuenciación, interpretación y limitacionesGracias a la next-generation secuencing (NGS) se han conseguido avances importantes en la comprensión de las bases genéticas de la MCD familiar en la que hay implicada una gran heterogeneidad de genes, así como mutaciones dentro de un mismo gen que son capaces de provocar el mismo fenotipo5,6. Así pues, es posible identificar la mutación casual de MCD en el 30-40% de los casos esporádicos y hasta en el 60-80% de los casos familiares7. Sin embargo, a pesar de los beneficios de esta nueva tecnología, se ha de ser muy cuidadoso en la interpretación y la traslación clínica de variantes halladas en el estudio. Se pueden encontrar mutaciones no frecuentes en la población general, pero es necesario distinguir si se trata de una mutación patogénica o una variante rara «benigna». Cuando la mutación en concreto se segrega con el fenotipo de otras familias con la misma mutación, hay pocas dudas de que se trate de una mutación patogénica. Sin embargo, cuando la mutación es «privada» (aparece por primera vez en una familia) la interpretación de esta se torna compleja y es probablemente uno de los mayores inconvenientes de esta nueva técnica. Para aclarar la patogenicidad de estas variantes es útil tener en cuenta la posible alteración que origina en la proteína. Así pues, las mutaciones que producen proteínas truncadas tienen más probabilidad de ser patogénicas; sin embargo, las mutaciones que generan el cambio de un aminoácido por otro generan mayor incertidumbre. Existen diferentes métodos que ayudan a determinar, pues, la significación de estas mutaciones. Se pueden hacer estudios in vitro sobre la repercusión en la proteína o bien utilizar programas bioinformáticos para analizar la conservación del aminoácido mutado a lo largo de las especies, determinando así si se encuentra en un dominio funcional. De manera paralela al desarrollo de las nuevas técnicas de secuenciación se están creando bases de datos con el genoma de algunas poblaciones bastante numerosas, por lo que si la mutación hallada es poco frecuente en la población referente existen más probabilidades de que sea patogénica. Un ejemplo sería la «dbSNP137common» (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/snp137Common.sql) en la que las variantes recogidas están presentes en más del 1% de la población. A pesar de estas herramientas, hay veces en las que no es posible determinar la significación de la mutación, denominándose entonces «variante de significado incierto»8.
Búsqueda de la significación clínica: del genenotipo al fenotipoHasta el momento han sido pocos y con cohortes escasas los estudios que han profundizado en las bases genéticas y en la correlación genotipo-fenotipo de la MCD familiar. Recientemente se han publicado los resultados del proyecto europeo INHERITANCE que ha incluido a 639 pacientes con MCD esporádica o familiar demostrada reclutados entre 2009 y 2011 en 8 centros clínicos europeos en los que se han investigado sistemáticamente mediante NGS no solo genes relacionados previamente con la MCD sino también aquellos que habían sido relacionados hasta el momento únicamente con otras miocardiopatías9. Los autores clasificaron las variantes encontradas en función de su patogenicidad, ayudándose de sistemas bioinformáticos en los casos necesarios, variantes relacionadas conocidas y relacionadas con la enfermedad, variantes probablemente relacionadas con la enfermedad y variantes potencialmente relacionadas con la enfermedad10. Los genes se agruparon según la función de la proteína que codificaban: canales iónicos, nuclear, membrana celular, sarcoméricos, del citoesqueleto y del disco intercalado.
En cuanto a los resultados, se trata de un estudio interesante, dado que el número de pacientes incluidos es considerable. Entre las características de los pacientes destaca que en su mayoría eran hombres (66%), el diámetro telediastólico medio del ventrículo izquierdo era de 64,4±11,2mm, la función ventricular media fue de 31,2±12,1% y se encontraban en su mayoría en clase funcional II o III de la NYHA (32% para cada una de estas clases). En cuanto a los resultados del test genético, el 46% (294) de todos los pacientes eran portadores de una mutación conocida y relacionada con la enfermedad y el 23% de los pacientes eran portadores de mutaciones con alta probabilidad de estar relacionadas con la enfermedad. Finalmente, se encontraron 141 variantes únicas potencialmente relacionadas con la enfermedad en 221 pacientes.
El mayor número de mutaciones conocidas de miocardiopatía se encontraron en PKP2, MYBPC3, DSP, DSC2 y SCN5A, las 5 primeras por orden de frecuencia. Sin embargo, al incluirse también las variantes de significado incierto (probable o potencialmente relacionadas con la enfermedad) los 5 genes con afectación más frecuente fueron TTN, PKP2, MYBPC3, DSP y RYR2.
Un dato destacable es que el 12,8% de los pacientes eran portadores de al menos 2 mutaciones conocidas de enfermedad. Los autores a continuación consultaron los resultados encontrados en la HGMD database y corroboraron que un amplio porcentaje de las mutaciones relacionadas con la MCD familiar estaban también descritas y relacionadas con la miocardiopatía arritmogénica del ventrículo derecho (31%), la miocardiopatía hipertrófica (16%) y canalopatías (6%). Por tanto, el solapamiento en las bases genéticas de las miocardiopatías es considerable.
La siguiente pregunta lógica que viene a continuación es si existe asociación específica entre el genotipo y el fenotipo. Así pues, se han encontrado diferencias estadísticamente significativas en los siguientes casos: el gen con mayor asociación con ser portador de DAI es RBM20; un mayor diámetro diastólico ventricular izquierdo se ha asociado con la mutación en TBX20; peor función ventricular con SYMD1 y CRYAB. Se encontró asociación significativa entre ser portador de mutaciones en MYPN y ser receptor de trasplante cardíaco.
Son pocos los estudios de correlación entre genotipo-fenotipo publicados hasta el momento. Destaca el realizado por Waldmüller en el que se secuenciaron los genes MYBPC3 y MYH7 en pacientes con miocardiopatía dilatada e hipertrófica y se encontró correlación entre algunas mutaciones en MYH7 y un mayor grado de regurgitación de la válvula mitral, tanto para pacientes con MCH como MCD11.
Estudios consistentes12 ya han puesto de manifiesto el pronóstico sombrío de los portadores de determinadas mutaciones en el gen LMNA, relacionado con arritmias ventriculares y muerte súbita. Mutaciones en el gen RBM20 (regulador del splicing del ARN premensajero, altamente expresado en el tejido cardíaco) se han relacionado así mismo con mayor incidencia de TV y antecedentes familiares de muerte súbita cardíaca13 y, como se ha mencionado anteriormente, con ser portador de DAI. Sin embargo, Refaat et al. no encontraron diferencias significativas entre ser portador de una mutación en RBM20 y una peor supervivencia, mayor tasa de trasplante ni de ser portador de DAI14.
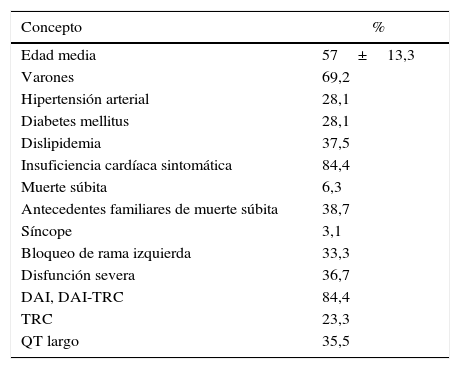
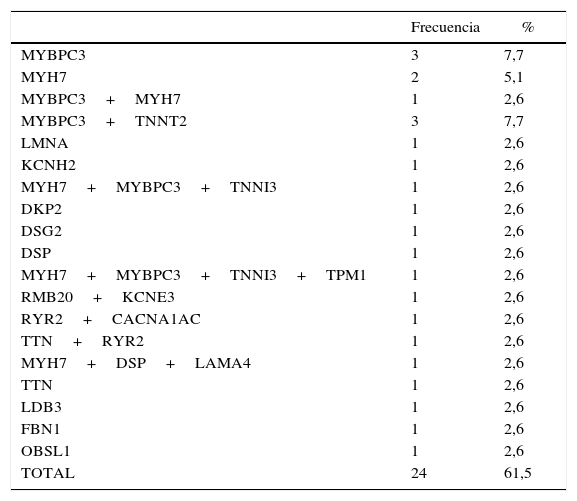
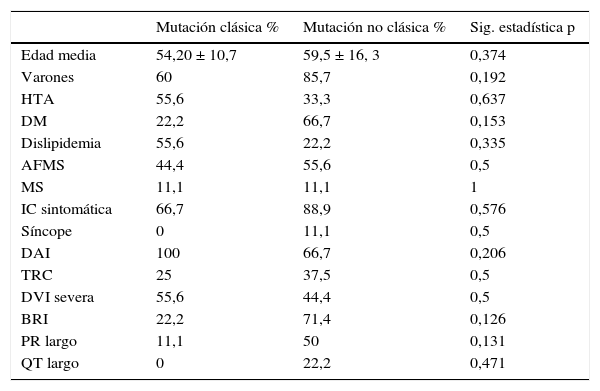
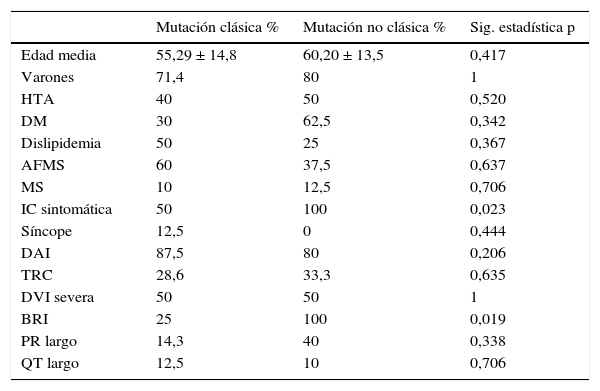
Nuestro grupo ha realizado recientemente un estudio de cohorte retrospectivo y ha estudiado todos los casos índices con diagnóstico de MCD familiar en seguimiento en consulta específica de cardiopatías familiares. Se planteó el objetivo de conocer si existía relación entre las características clínicas de los pacientes y los resultados del test genético. Para ello se compararon los pacientes con mutaciones simples frente a aquellos con mutaciones múltiples (2 o más mutaciones), así como los pacientes con mutaciones clásicas (reportadas en mayor grado de frecuencia hasta la llegada del método NGS: MYH7, MYBPC3, TNNT2, TNNI3, TPM1 y LMNA) frente a aquellos con mutaciones no clásicas (el resto de mutaciones relacionadas con la enfermedad que no están incluidas en las clásicas). Para lo cual se analizaron los 39 casos índice consecutivos desde la apertura de la consulta en 2009 hasta marzo de 2014. Se recogieron los resultados del estudio genético, así como las características clínicas de los pacientes. En cuanto a los resultados, destacan entre las características clínicas que el 6,3% presentaron muerte súbita, el 38,7% antecedentes familiares de muerte súbita, el 84,4% insuficiencia cardíaca (DVI severa un 36,7%) y un 3,1% síncope. El 84,4% eran portadores de DAI o DAI-TRC y el 23,3% de TRC, presentando BRI un 33,3% y QT largo un 35,5% (tabla 1). Los resultados del estudio genético se recogen en la tabla 2. Presentaron mutación genética un 61,5%, siendo mutaciones múltiples un 41,7% y de genes no clásicos un 58,3%. No existieron diferencias clínicas significativas entre portadores de mutaciones de genes clásicos y no clásicos (tabla 3); existió en los portadores de mutaciones múltiples mayor porcentaje de insuficiencia cardíaca (100 vs. 50%; p=0,023) y mayor porcentaje de BRI en los portadores de más de 2 mutaciones (100 vs. 25%; p=0,019) (tabla 4).
| Concepto | % |
|---|---|
| Edad media | 57±13,3 |
| Varones | 69,2 |
| Hipertensión arterial | 28,1 |
| Diabetes mellitus | 28,1 |
| Dislipidemia | 37,5 |
| Insuficiencia cardíaca sintomática | 84,4 |
| Muerte súbita | 6,3 |
| Antecedentes familiares de muerte súbita | 38,7 |
| Síncope | 3,1 |
| Bloqueo de rama izquierda | 33,3 |
| Disfunción severa | 36,7 |
| DAI, DAI-TRC | 84,4 |
| TRC | 23,3 |
| QT largo | 35,5 |
| Frecuencia | % | |
|---|---|---|
| MYBPC3 | 3 | 7,7 |
| MYH7 | 2 | 5,1 |
| MYBPC3+MYH7 | 1 | 2,6 |
| MYBPC3+TNNT2 | 3 | 7,7 |
| LMNA | 1 | 2,6 |
| KCNH2 | 1 | 2,6 |
| MYH7+MYBPC3+TNNI3 | 1 | 2,6 |
| DKP2 | 1 | 2,6 |
| DSG2 | 1 | 2,6 |
| DSP | 1 | 2,6 |
| MYH7+MYBPC3+TNNI3+TPM1 | 1 | 2,6 |
| RMB20+KCNE3 | 1 | 2,6 |
| RYR2+CACNA1AC | 1 | 2,6 |
| TTN+RYR2 | 1 | 2,6 |
| MYH7+DSP+LAMA4 | 1 | 2,6 |
| TTN | 1 | 2,6 |
| LDB3 | 1 | 2,6 |
| FBN1 | 1 | 2,6 |
| OBSL1 | 1 | 2,6 |
| TOTAL | 24 | 61,5 |
| Mutación clásica % | Mutación no clásica % | Sig. estadística p | |
|---|---|---|---|
| Edad media | 54,20 ± 10,7 | 59,5 ± 16, 3 | 0,374 |
| Varones | 60 | 85,7 | 0,192 |
| HTA | 55,6 | 33,3 | 0,637 |
| DM | 22,2 | 66,7 | 0,153 |
| Dislipidemia | 55,6 | 22,2 | 0,335 |
| AFMS | 44,4 | 55,6 | 0,5 |
| MS | 11,1 | 11,1 | 1 |
| IC sintomática | 66,7 | 88,9 | 0,576 |
| Síncope | 0 | 11,1 | 0,5 |
| DAI | 100 | 66,7 | 0,206 |
| TRC | 25 | 37,5 | 0,5 |
| DVI severa | 55,6 | 44,4 | 0,5 |
| BRI | 22,2 | 71,4 | 0,126 |
| PR largo | 11,1 | 50 | 0,131 |
| QT largo | 0 | 22,2 | 0,471 |
| Mutación clásica % | Mutación no clásica % | Sig. estadística p | |
|---|---|---|---|
| Edad media | 55,29 ± 14,8 | 60,20 ± 13,5 | 0,417 |
| Varones | 71,4 | 80 | 1 |
| HTA | 40 | 50 | 0,520 |
| DM | 30 | 62,5 | 0,342 |
| Dislipidemia | 50 | 25 | 0,367 |
| AFMS | 60 | 37,5 | 0,637 |
| MS | 10 | 12,5 | 0,706 |
| IC sintomática | 50 | 100 | 0,023 |
| Síncope | 12,5 | 0 | 0,444 |
| DAI | 87,5 | 80 | 0,206 |
| TRC | 28,6 | 33,3 | 0,635 |
| DVI severa | 50 | 50 | 1 |
| BRI | 25 | 100 | 0,019 |
| PR largo | 14,3 | 40 | 0,338 |
| QT largo | 12,5 | 10 | 0,706 |
¿Cuándo solicitamos un estudio genético en un paciente con MCD? La ESC sostiene una serie de recomendaciones para la realización de test genéticos, pero están destinados a todas las miocardiopatías en general15, por lo que en el caso de la MCD, en el que la variabilidad en la expresión fenotípica y la heterogenicidad genética es tan amplia, no existe una recomendación específica. El estudio genético estaría destinado a aquellos pacientes con MCD idiopática, es decir, en aquellos en los que se hayan descartado todas las posibles causas de miocardiopatía dilatada, salvo la genética. En los casos en los que haya antecedentes familiares de miocardiopatía, insuficiencia cardíaca o muerte súbita de causa no filiada, la sospecha de que estamos ante un caso índice de miocardipatía es alta y el estudio genético podría confirmar esa asociación familiar. Pero debido a que la rentabilidad genética oscila entre el 60 y el 80%, un resultado negativo no descartaría la causa genética. En los casos esporádicos, así como en aquellos con características fenotípicas borderline también aportaría información, y podría llevar a un diagnóstico definitivo, aunque existe controversia sobre si se debe realizar a todos los pacientes de manera sistemática, teniendo en cuenta además que es relativamente costoso y no está disponible en todos los centros.
Las utilidades actualmente están más dirigidas al manejo de los familiares que al del propio caso índice, ya que conocer la mutación exacta que porta el caso índice una vez se ha expresado la enfermedad no hará variar en la mayoría de los casos ni el tratamiento ni el pronóstico, al menos hasta que los estudios que analizan la correlación fenotipo-genotipo sean consistentes y existan recomendaciones de las sociedades científicas al respecto. Pero conocer la mutación del caso índice nos permite hacer un screening genético en sus familiares, de manera que aquellos que no son portadores de la mutación pueden cesar en el seguimiento con el ahorro de coste psicológico derivado de la incertidumbre de presentar la enfermedad en el futuro y de coste económico para el sistema, ya que estos pacientes con un seguimiento tradicional según las recomendaciones de la ECS deben acudir a revisiones sistemáticas desde la infancia y durante el resto de su vida, con una periodicidad variable en función de la franja etaria (cada 2-5 años), con realización de pruebas complementarias (ECG, ecocardiografía y Holter de 24h)15,16. La utilidad en los portadores sanos proviene de un seguimiento más estrecho para detectar con precocidad la aparición de la clínica que permita cambios en el estilo de vida y la instauración precoz de tratamiento y de DAI en caso de mutaciones con alto riesgo de muerte súbita como las descritas en la LMNA. En el caso de la MCD no existen estudios en nuestro medio que hayan analizado el impacto clínico y económico, pero sí existe un estudio realizado por Cobo-Martos et al. en el que se analizó este impacto en el caso de la MCH y se concluyó que el coste directo de realizar el estudio genético (coste del análisis genético menos el ahorro sanitario) era de 135 euros por familia analizada17. Resultaría interesante realizar un estudio similar en el caso de la MCD familiar.
Otra de las utilidades indiscutibles de la realización de test genéticos deriva de permitir un consejo genético que incluye el reproductivo, el profesional y el deportivo, que debe ser aportado por profesionales experimentados en la materia. Se les deberá explicar los riesgos y beneficios del test y sus implicaciones clínicas, psicológicas, sociales, laborales y deportivas, tanto del resultado positivo como del negativo. Idealmente el consejo genético debería ser aportado antes de la gestación, explicándose las posibles alternativas reproductivas.
En el futuro, la realización de estudios de base poblacional que analicen la posible asociación entre genotipo y fenotipo podría ayudar a individualizar mejor el tratamiento de estos pacientes en función de las posibles diferencias en cuanto a su expresión clínica, así como conocer si existen diferencias pronósticas en la evolución en función de los resultados en el estudio genético.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

