La ventana aorto-pulmonar es una rara anomalía cardiaca congénita; sin embargo, no es infrecuente que se asocie a diferentes cardiopatías congénitas. La comunicación interauricular es la anomalía más frecuentemente asociada. La combinación de ventana aorto-pulmonar e interrupción del arco aórtico constituye el prototipo donde la ventana aorto-pulmonar se asocia a una cardiopatía congénita compleja. Estas asociaciones de ventana aorto-pulmonar con otras cardiopatías congénitas complejas pueden ser muy variadas e inusuales, aumentando los riesgos de morbimortalidad en la evolución de los pacientes que las padecen. Presentamos 3 casos clínicos donde la ventana aorto-pulmonar se asocia a otras cardiopatías complejas tales como interrupción del arco aórtico, obstrucción del tracto de salida ventricular izquierdo y comunicación interventricular múltiple. Estas asociaciones complejas pueden ser corregidas quirúrgicamente en periodo neonatal con unos resultados aceptables, a pesar de su rareza y escasa experiencia quirúrgica acumulada.
The aorto-pulmonary window is a rare congenital heart anomaly, however, this anomaly is not uncommon associated with different congenital heart diseases. Atrial septal defect is the most frequently associated anomaly. The combination of aorto-pulmonary window and interrupted aortic arch constitutes the prototype which the aorto-pulmonary window is associated with complex congenital heart disease. These aorto-pulmonary window associations with other complex congenital heart diseases can be very varied and unusual, increasing the risk of morbidity and mortality in the evolution of patients who suffer from them. We present three case reports where the aorto-pulmonary window is associated with other complex heart diseases such as interrupted aortic arch, left ventricular outflow tract obstruction, and multiple ventricular septal defects. These complex associations can be surgically corrected in neonatal period with acceptable results, despite their rarity and little accumulated surgical experience.
La ventana aorto-pulmonar (VAP) es una rara anomalía cardiaca congénita, cuya incidencia se sitúa en torno al 0,15% del total de pacientes diagnosticados de cardiopatía congénita1. A pesar de su rareza, no es infrecuente que se asocie a diferentes anomalías cardiacas congénitas (50%)2,3 tales como comunicación interventricular (CIV), comunicación interauricular (CIA), tetralogía de Fallot, interrupción del arco aórtico (IAAo), transposición de grandes arterias,… La asociación de la VAP y la IAAo es una asociación característica que se describe en las diferentes series sobre la VAP publicadas en la literatura4.
Casos clínicosDurante los últimos 10 años se han intervenido en nuestro centro 8 pacientes con diagnóstico de VAP, rango de edad entre 5 días/3 meses y un peso entre 2,2-4,3kg. Cuatro de ellos con VAP asociada a otras anomalías cardiacas: 2 CIA, una IAAo, una CIV múltiples con CIA. Un caso intervenido de VAP sin otros defectos asociados en el momento de la intervención inicial, desarrolló posteriormente una obstrucción subaórtica severa compleja. Procedemos a describir resumidamente estos 3 casos por su infrecuencia.
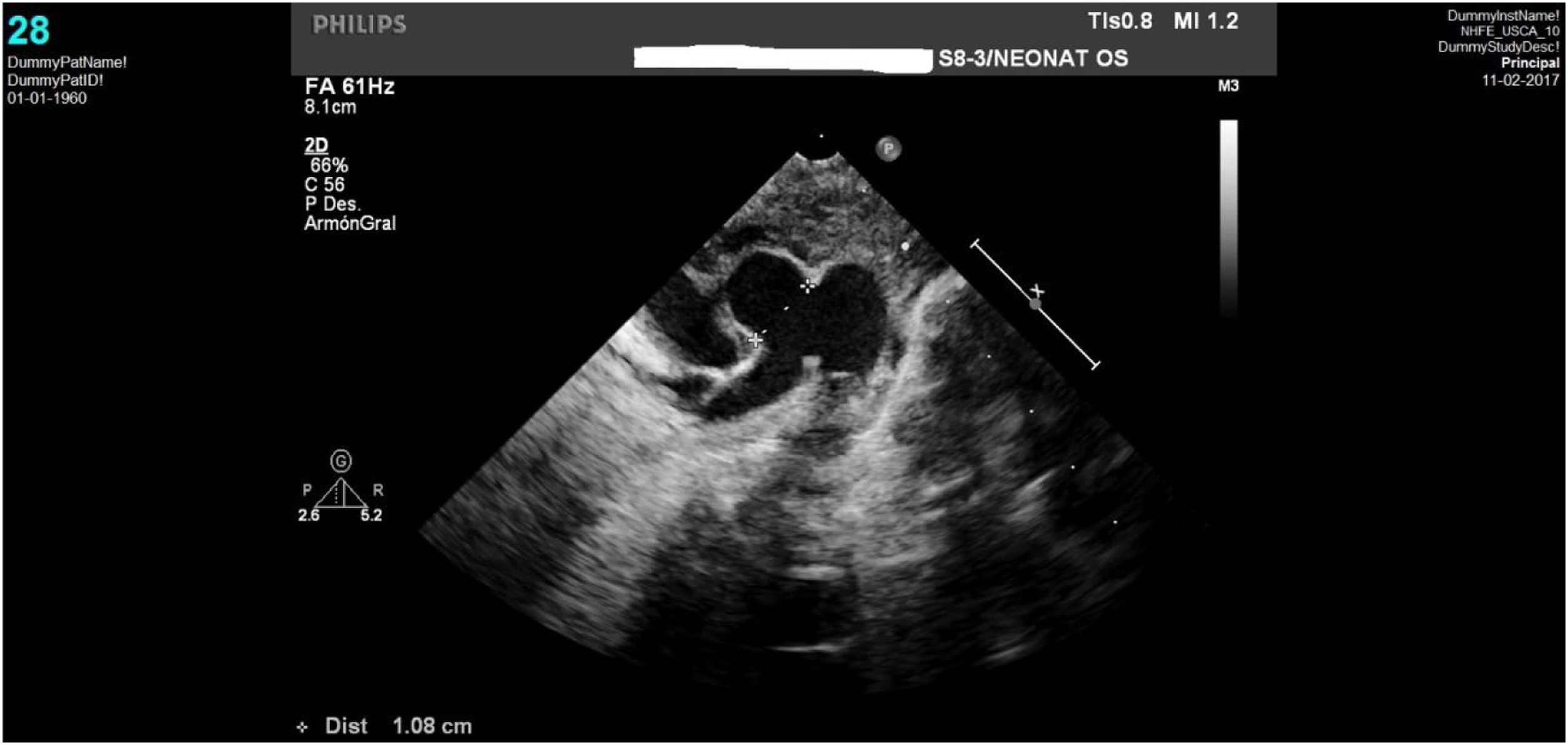
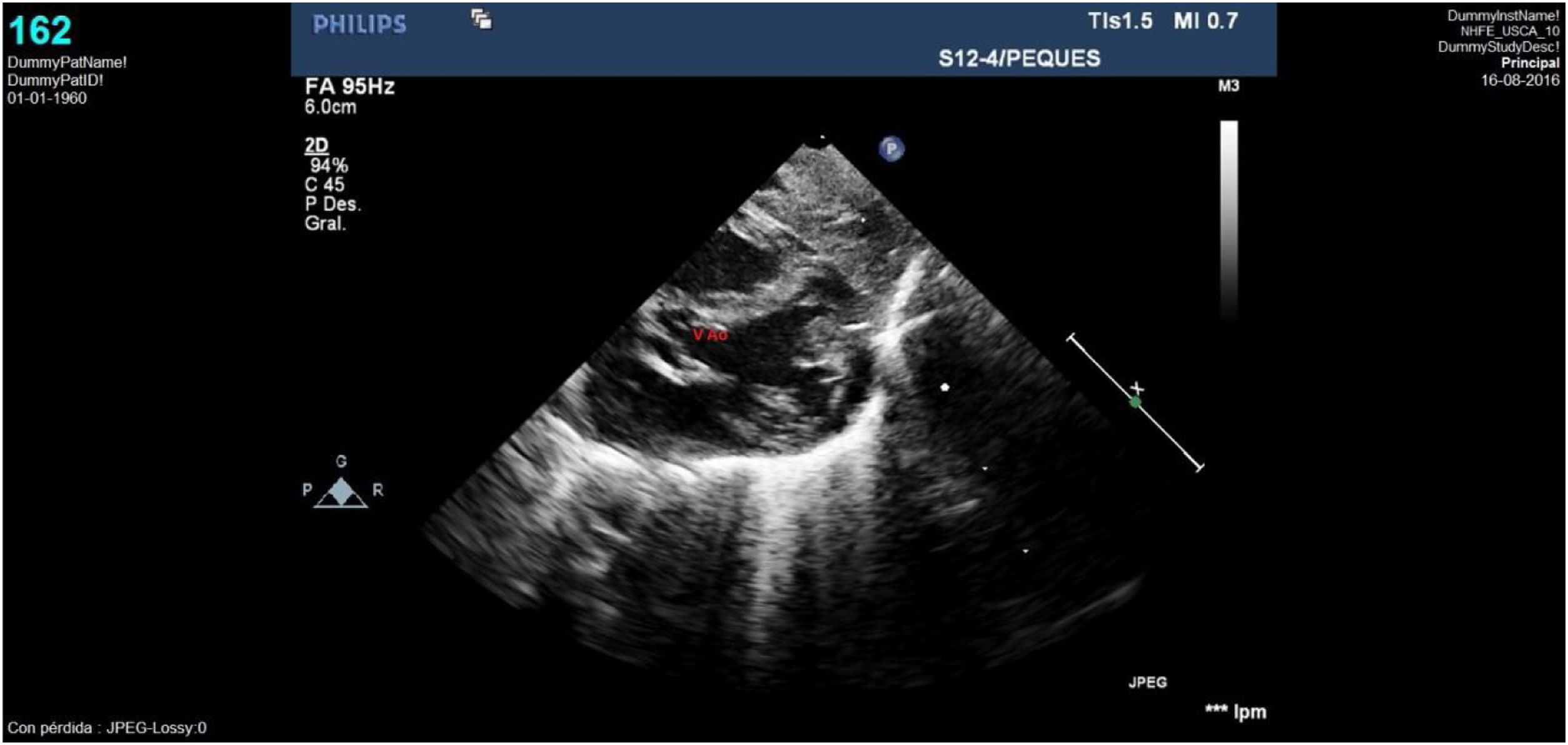
Caso clínico 1Neonata de 2,7kg donde la ecocardiografía (figs. 1 y 2) mostraba una VAP de unos 10mm (fig. 1); el arco aórtico mostraba una salida muy próxima de los 3 troncos supraaórticos y una interrupción del mismo tras la salida de la arteria subclavia izquierda (IAAo tipo A) (fig. 2), existiendo un ductus amplio que se continuaba con la aorta torácica descendente; se identificaba vena cava superior izquierda (VCSI).
.")
. Se identifica además, la amplia ventana aórtica tipo II.")
Se procedió a su reparación completa, cerclándose ambas ramas pulmonares al iniciarse la CEC. La perfusión del hemicuerpo distal se aseguró a través de la VAP y del ductus sistémico. Alcanzados los 22°C se seccionó el ductus arterioso, se avanzó la cánula aórtica al tronco braquiocefálio derecho, iniciándose perfusión cerebral selectiva, procediéndose a la reparación del arco aórtico mediante avance de la aorta torácica descendente al suelo del arco proximal y aorta ascendente distal. Una vez reparado el arco, se recolocó la cánula arterial de perfusión en aorta ascendente distal reperfundiéndose la totalidad del cuerpo. Tras dosis de cardioplejía de mantenimiento, aprovechando la fase de by-pass de calentamiento, se procedió a la reparación de la VAP vía transaórtica.
El postoperatorio transcurrió sin incidencias, con controles ecocardiográficos satisfactorios y la paciente fue dada de alta hospitalaria al 11.° día de postoperatorio. A los 23 meses de vida se detectó una recoartación moderada con amortiguación de la curva de pulsatilidad en aorta abdominal por lo que se realizó una angioplastia con buen resultado angiográfico y hemodinámico.
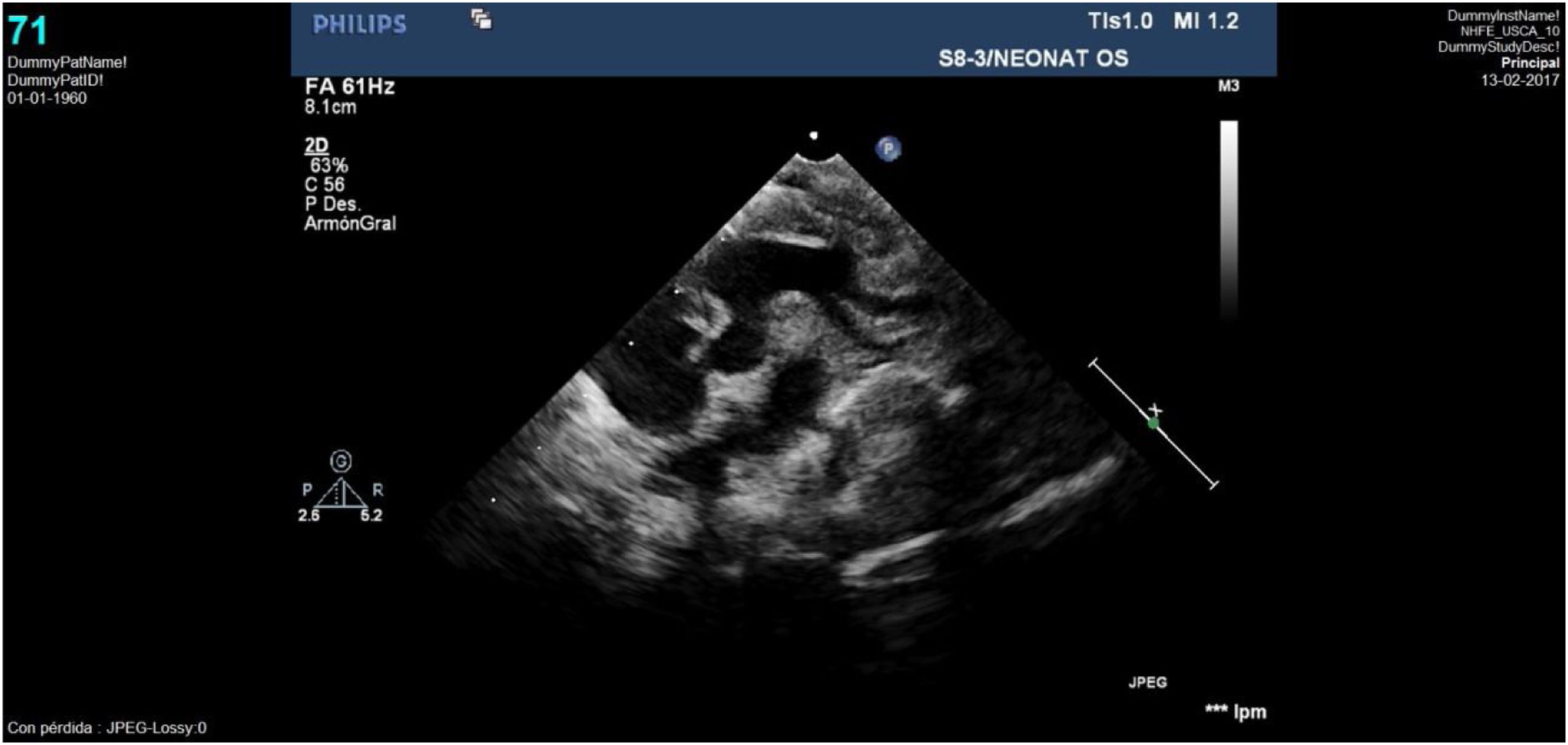
Caso clínico 2Varón prematuro de 35 semanas y retraso del crecimiento intrauterino (peso al nacimiento de 1.590g; p<3). La ecocardiografía (figs. 3-4) mostró una VAP amplia cercana al plano valvular y proximal a la salida de las ramas pulmonares, anillo aórtico de 4,6mm y pulmonar de 6,7mm, sin datos de obstrucción en tractos de salida. Así mismo, se apreciaba probable ausencia de vena cava superior derecha con seno coronario dilatado donde drenaba una VCSI. El arco aórtico distal era uniformemente pequeño sin datos de coartación, y sin apreciar otros hallazgos de significación patológica.


Debido al bajo peso al nacimiento, se decidió retrasar el momento de la intervención quirúrgica con objeto de conseguir una ganancia ponderal favorable. Sin embargo, la situación clínica del paciente obligó con apenas un mes de vida y un peso de 2,2kg a proceder a su reparación. Se intervino bajo CEC e hipotermia moderada, reparándose vía transaórtica la VAP.
El postoperatorio transcurrió sin incidencias, procediéndose al 13.° día del postoperatorio al alta hospitalaria, siendo los controles ecocardiográficos iniciales de seguimiento normales. Sin embargo, a partir del 5.° mes de vida, comenzó a detectarse en los controles ecocardiográficos una estenosis subvalvular aórtica de grado ligero-moderado atribuible a una membrana subaórtica completa. Dicha obstrucción fue progresando rápidamente, de modo que a los 11 meses la ecocardiografía mostró una estenosis aórtica subvalvular severa atribuible a dicha membrana; por lo que se reintervino resecando dicha membrana con buen resultado anatómico y hemodinámico.
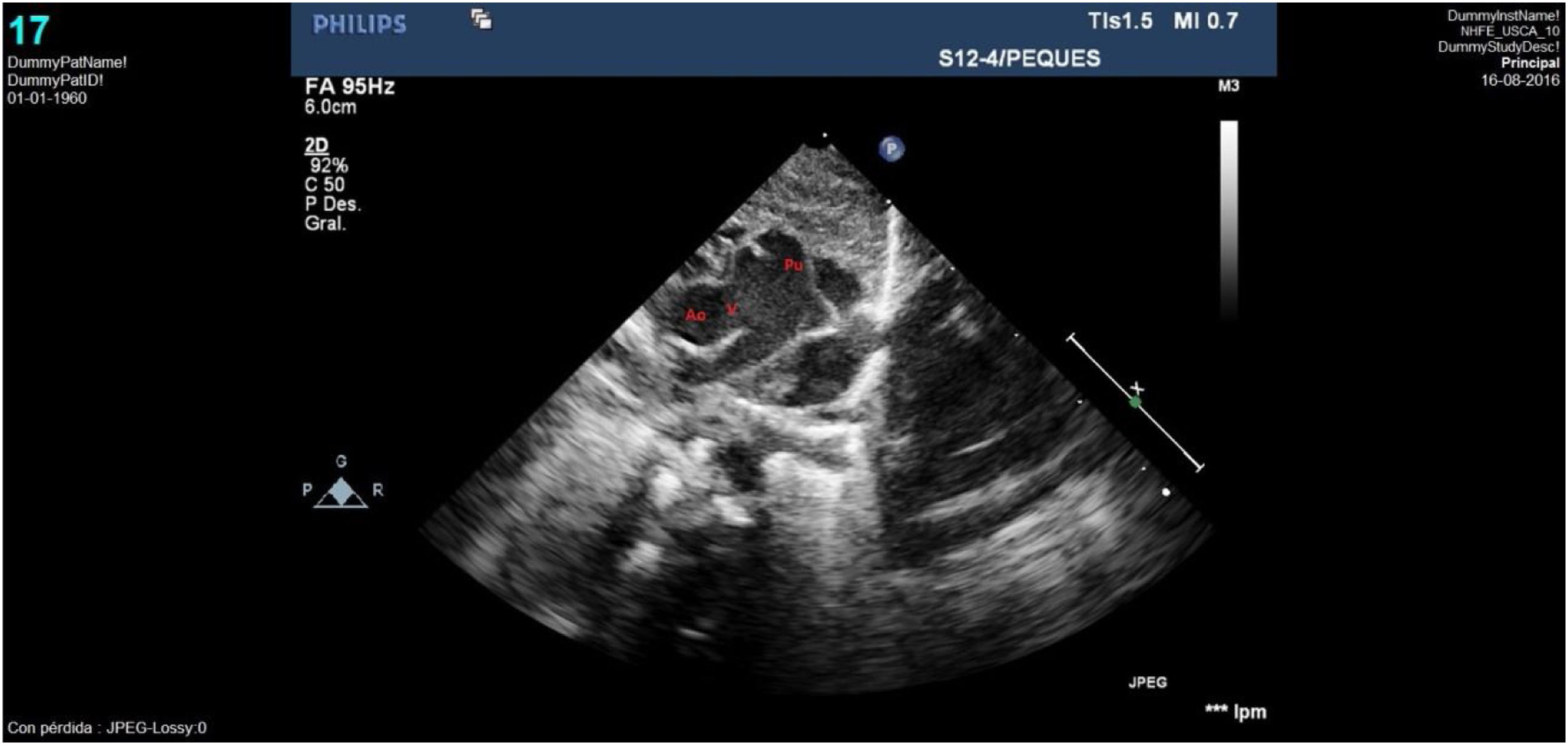
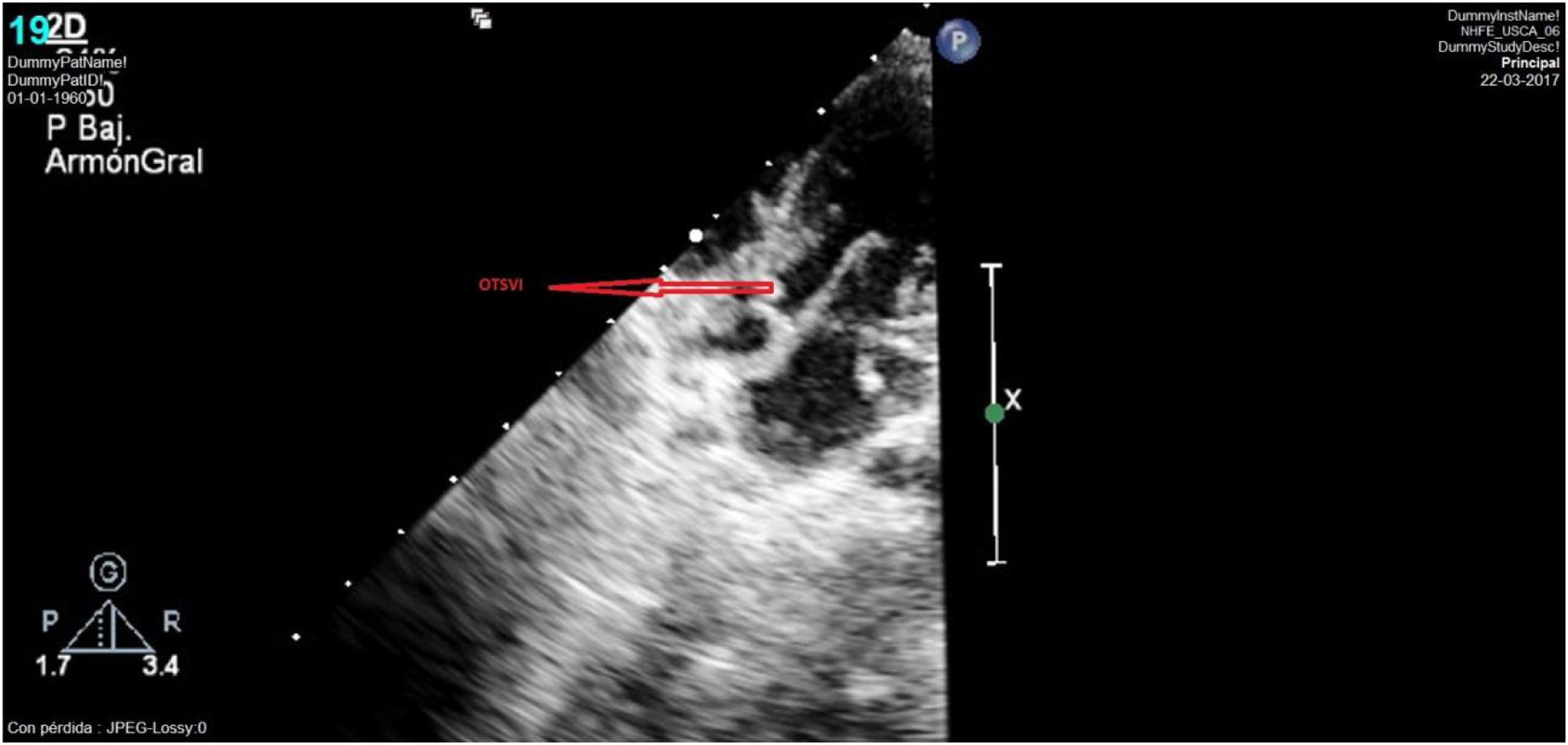
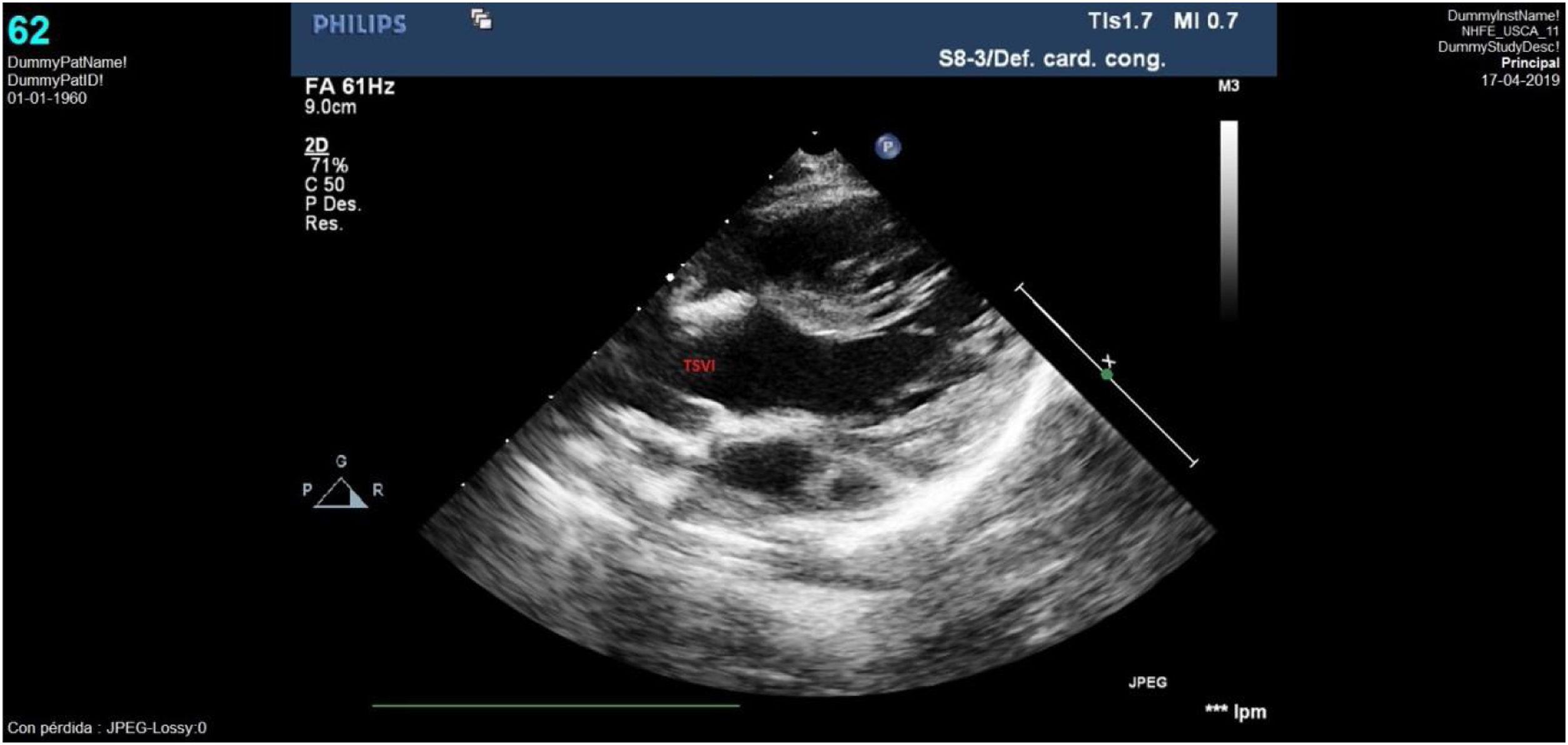
El seguimiento posterior puso de manifiesto un tracto de salida ventricular izquierdo (TSVI) que progresivamente fue quedando pequeño respecto al crecimiento del paciente. Así, la ecocardiografía realizada a los 30 meses de vida evidenció un TSVI tuneliforme y angosto de unos 5mm con un anillo aórtico hipoplásico de 6mm (Z-score: −4) y gradiente pico de 100mmHg; anillo pulmonar de 12mm con válvula normofuncionante y ventrículo izquierdo hipertrófico con buena función (fig. 5). Con esta situación ecocardiográfica, aunque el paciente se encontraba clínicamente asintomático, se decidió plantear nueva intervención quirúrgica para solventar la obstrucción del TSVI, realizándose una operación de Ross-Konno con buen resultado (fig. 6). Actualmente el paciente se encuentra asintomático, con autoinjerto pulmonar en posición aórtica normofuncionante y conducto VD-AP ligeramente obstructivo, sin insuficiencia valvular.


Neonato de 2,6kg que presentaba una VAP asociada a 2 CIV amplias, una de localización perimembranosa y otra de localización en septo muscular liso por encima de la banda moderadora; así mismo, presentaba una VCSI a seno coronario.
Se intervino bajo CEC procediéndose a una reparación completa mediante cierre transauricular/transtricuspídeo de ambas CIV y reparación transaórtica de la VAP. Así mismo, se cerró un pequeño ductus permeable y se constató intraoperatoriamente la existencia de un arco aórtico derecho y una pequeña VCSI con presencia de vena innominada. El postoperatorio transcurrió sin incidencias, siendo dado de alta hospitalaria al 15.° día del postoperatorio. Actualmente el paciente se encuentra asintomático sin defectos residuales.
DiscusiónLa VAP es una rara anomalía cardiaca congénita, con una incidencia entre el 0,15 y el 0,2% del total de cardiopatías congénitas diagnosticadas1,5; en nuestro centro las cifras han sido similares, y en los últimos, casi 10 años, se han intervenido 8 VAP sobre un total de 1.381 procedimientos quirúrgicos mayores realizados en cirugía cardiaca infantil. Sin embargo, es frecuente (50-52%)2,3,5 que la VAP se asocie a otras anomalías cardiacas congénitas que pueden oscilar desde la CIA, que es la asociación más frecuente con hasta un 65% de los casos5 (en nuestra serie 3 pacientes presentaron CIA asociada), pasando por una asociación muy característica en la literatura3–6, pero poco frecuente (3,5-4,2%)5,6 como es la IAAo (un caso en nuestra serie) o llegando a asociaciones muy infrecuentes en la literatura como en los casos presentados de reparación neonatal de la VAP con posterior desarrollo meses después de una obstrucción severa del TSVI o el caso clínico 3 donde a la presencia de una VAP se sumaban la existencia de 2 CIV amplias y una CIA.
Si bien la corrección quirúrgica de la VAP aislada muestra unos resultados favorables a pesar de su escasa frecuencia, cuando se asocia a otras enfermedades cardiacas congénitas complejas, estas asociaciones pueden resultar en un incremento significativo de la morbimortalidad a corto y largo plazo debido a la escasez de experiencia acumulada por los grupos quirúrgicos y en general en la literatura, donde con frecuencia o bien las series son muy limitadas en el número de casos o incluso para algunas asociaciones complejas la literatura se reduce a casos clínicos aislados3–6.
Las anomalías del retorno venoso sistémico se describen con cierta frecuencia asociadas a la IAAo y a la VAP5. En nuestros 3 casos presentados estas anomalías venosas sistémicas estuvieron siempre presentes y consistieron en la persistencia de la VCSI. Esta persistencia de la VCSI no solo estuvo presente en el caso de la VAP asociada a IAAo, sino que también se dio en los casos clínicos 2 y 3 donde no existía IAAo. Incluso en el caso clínico 2 la anomalía venosa sistémica consistió en una ausencia de la vena cava superior derecha con una vena innominada que conectaba con la VCSI que drenaba en el seno coronario. Estas anomalías venosas sistémicas no tuvieron repercusión quirúrgica significativa, ni a nuestro juicio complicaron el desarrollo de las intervenciones quirúrgicas, ya que en los casos 1 y 2 no fue preciso realizar una atriotomía derecha, recogiéndose adecuadamente el retorno venoso sistémico con una cánula alojada en la aurícula derecha y otra en la vena cava inferior. En el caso clínico 3 donde fue preciso realizar una atriotomía derecha para el cierre de las CIV, se cercló temporalmente la VCSI al existir afortunadamente vena innominada que conectaba la VCSI con la cava superior derecha, canulándose ambas cavas para recoger el retorno venoso sistémico.
La morbimortalidad precoz tras la reparación quirúrgica de la VAP aislada es baja, pero esta se incrementa significativamente cuando la VAP se asocia a otras lesiones cardiacas, especialmente a la IAAo3. En este incremento de morbimortalidad parece influir la menor experiencia quirúrgica acumulada por los diferentes grupos debido a la infrecuencia de esas asociaciones complejas de la VAP3–6. Pero, también puede tener influencia el bajo peso que muestran los pacientes de las series presentadas de la VAP y la IAAo en la literatura (serie Rouberti et al. con peso medio de 2,6kg3. Serie de Melbourne con peso medio inferior a 3kg6). En nuestra serie, la paciente con la VAP y la IAAo presentaba en el momento de la intervención un peso de 2,7k,; pero esta característica de peso inferior a 3kg fue también extensible tanto al caso 3 como especialmente al caso 2 donde a su bajo peso al nacimiento, secundario a la prematuridad y el retraso de crecimiento intrauterino, se unió la rápida aparición de clínica derivada de la sobrecirculación pulmonar y malperfusión sistémica secundaria a la disminución de la resistencia vascular pulmonar con el paso del tiempo tras el nacimiento, lo que obligó a su intervención con 2,2kg y una edad de poco más de un mes de vida. Afortunadamente, la cirugía que precisó inicialmente consistió en un cierre quirúrgico de la VAP aislada, ya que posiblemente por su bajo peso inicial no había datos obstructivos a nivel del TSVI a pesar de su tamaño límite.
Es reconocido en la literatura que la asociación de la VAP y la IAAo no solo presenta un mayor riesgo de morbimortalidad precoz, sino que también presenta un mayor riesgo de morbilidad a largo plazo, especialmente en forma de obstrucción a nivel de la anastomosis aórtica y en menor medida en forma de estenosis de las arterias pulmonares3,5. En el caso clínico 1, la paciente precisó una angioplastia a los 23 meses de vida sobre la anastomosis del arco aórtico con buen resultado; en este caso tras una extensa movilización de la aorta torácica descendente y una resección de la totalidad del tejido ductal, animados por una anatomía más favorable al tratarse de una IAAo tipo A, procedimos a realizar una anastomosis T-L directa de la aorta torácica descendente sobre el arco aórtico proximal sin interponer parche de ampliación. Existe una amplia controversia sobre la forma de reparar la IAAo asociada o no a la VAP respecto a realizar avances aórticos directos vs. interposición de parches de ampliación en dicha anastomosis, siendo en las series históricas publicadas de IAAo con VAP más frecuente realizar avances aórticos directos5,6. Sin embargo, en los últimos casos de dichas series parece existir una mayor predisposición al empleo de parches de ampliación sobre esas anastomosis aórticas, especialmente en los casos de IAAo tipo B5. A este respecto, la técnica de parche único para corregir la IAAo asociada con la VAP publicada por Rouberti et al. parece ofrecer unos resultados prometedores con nula morbilidad sobre la anastomosis aórtica o sobre las ramas pulmonares en el seguimiento a largo plazo3.
La obstrucción del TSVI concomitante con la existencia de la VAP es una entidad muy infrecuente4,7. Si bien en el caso clínico 2 inicialmente no existían datos ecocardiográficos de obstrucción en el TSVI, pensamos que esto fue debido al bajo peso inicial del paciente, que a pesar de tener un anillo aórtico de tamaño límite inicialmente (4,6mm) no llegaba a producir un mismatch entre la válvula aórtica nativa y el tamaño del paciente. La falta de crecimiento del anillo aórtico unido a un TSVI de aspecto tuneliforme que tampoco se desarrollaron a pesar de la reintervención dirigida a la resección de la membrana subaórtica llegó a provocar finalmente con el crecimiento somático del paciente en los meses sucesivos un mismatch entre dicho TSVI (Z-score −4) y el tamaño del paciente, que obligó a la realización de una cirugía de Ross-Konno. La realización de una angio-TAC preoperatoria permitió ayudar a establecer la viabilidad del autoinjerto pulmonar previamente a la cirugía de Ross-Konno planificada, a pesar de la cirugía previa de cierre de la VAP practicada; creemos también que el abordaje transaórtico de la VAP pudo ayudar a preservar la utilidad del autoinjerto pulmonar.
En resumen, la VAP siendo una entidad rara, no es infrecuente que se presente asociada a otras formas complejas de cardiopatía congénita (3/8 casos en nuestra serie), pudiendo ser corregidas estas asociaciones complejas en periodo neonatal con unos resultados aceptables, a pesar de la escasa experiencia quirúrgica acumulada en la literatura.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.