


La epiteliopatía pigmentaria placoide multifocal posterior aguda es una entidad poco frecuente, autolimitada, de carácter inflamatorio a nivel de coriocapilaris y capas externas de la retina.
Caso clínicoPaciente varón de 26 años de edad, que acude por baja visual de inicio repentino en ambos ojos y 3 días de evolución, acompañada de fotopsias. A la exploración se encontró una capacidad visual de 20/200 en el ojo derecho y de 20/80 en el ojo izquierdo. No se encontró reacción inflamatoria en la cámara anterior ni tampoco vitritis en ningún ojo. En el polo posterior se encontraron lesiones de aspecto cremoso blanquecino subretinianas, en forma de placas. En la fluorangiografía retiniana se encontró hipofluorescencia en fases tempranas de las lesiones, vistas en fotos clínicas con hiperfluorescencia en fases tardías. En función de los datos clínicos y angiográficos se diagnosticó como epiteliopatía pigmentaria placoide multifocal posterior aguda.
ConclusionesLa epiteliopatía pigmentaria placoide multifocal posterior aguda es una entidad inflamatoria de origen desconocido que forma parte del diagnóstico diferencial, de las llamadas enfermedades placoideas de la retina.
Acute posterior multifocal placoid pigment epitheliopathy is an unusual, self-limited, inflammatory disease that affects the choriocapillaris, and external retinal layers.
Clinical caseA 26 year-old male patient complained of decreased visual acuity, as well as photopsia in both eyes for the past three days. Best corrected visual acuity was 20/200 in the right eye and 20/80 in the left eye. There was no anterior chamber inflammation or vitritis in either eye. There were plaque-like, cream-coloured sub-retinal lesions with ill-defined borders in the posterior pole of both eyes. Fluorescein angiography showed hypofluorescent lesions in early phases that corresponded to the lesions seen in the clinical examination. These lesions were hyperfluorescent in later phases of the angiography. Based on the clinical and angiographic findings, an acute posterior multifocal placoid pigment epitheliopathy diagnosis was made.
ConclusionsAcute posterior multifocal placoid pigment epitheliopathy is an inflammatory condition of unknown origin that is part of the differential diagnosis of placoid retinal diseases.
La epiteliopatía pigmentaria placoide multifocal posterior aguda fue descrita por primera vez en 1968 por Gass1. En este reporte se detalló la presencia de lesiones multifocales placoideas (en forma de placa) a nivel de las capas externas de la retina y del epitelio pigmentado, en 3 mujeres por lo demás sanas. Hasta la fecha continúa la polémica sobre la etiología y localización precisa en el espesor retinocoroideo de estas lesiones2,3.
En la mayoría de los casos esta entidad es benigna y autolimitada y no requiere de intervención mayor por parte del médico tratante, como los casos que reportó Gass inicialmente1,4.
Las manifestaciones clínicas de epiteliopatía pigmentaria placoide multifocal posterior aguda se presentan con pérdida súbita de la visión central, que describe el paciente como: visión borrosa, escotoma paracentral, metamorfopsia, «puntos» en el campo visual y fotopsias4.
En el 77% de los casos se presenta una visión inicial menor o igual a 20/25, mientras que en el 58% de los casos es menor o igual a 20/404.
La baja visual puede ser uni o bilateral (esta es la más frecuente, estando presente en el 75% de los casos)5. Si la forma de presentación es unilateral, el segundo ojo llega ocasionalmente a involucrarse después de algunos días o semanas. Pueden acompañar a los síntomas oculares: cefalea, rigidez en el cuello y ocasionalmente malestar general, así como algún antecedente de un síndrome viral o de vacunación recientes.
En cuanto a la epidemiología, tanto hombres como mujeres se afectan por igual y las edades de presentación son entre los 20 y 50 años4,5.
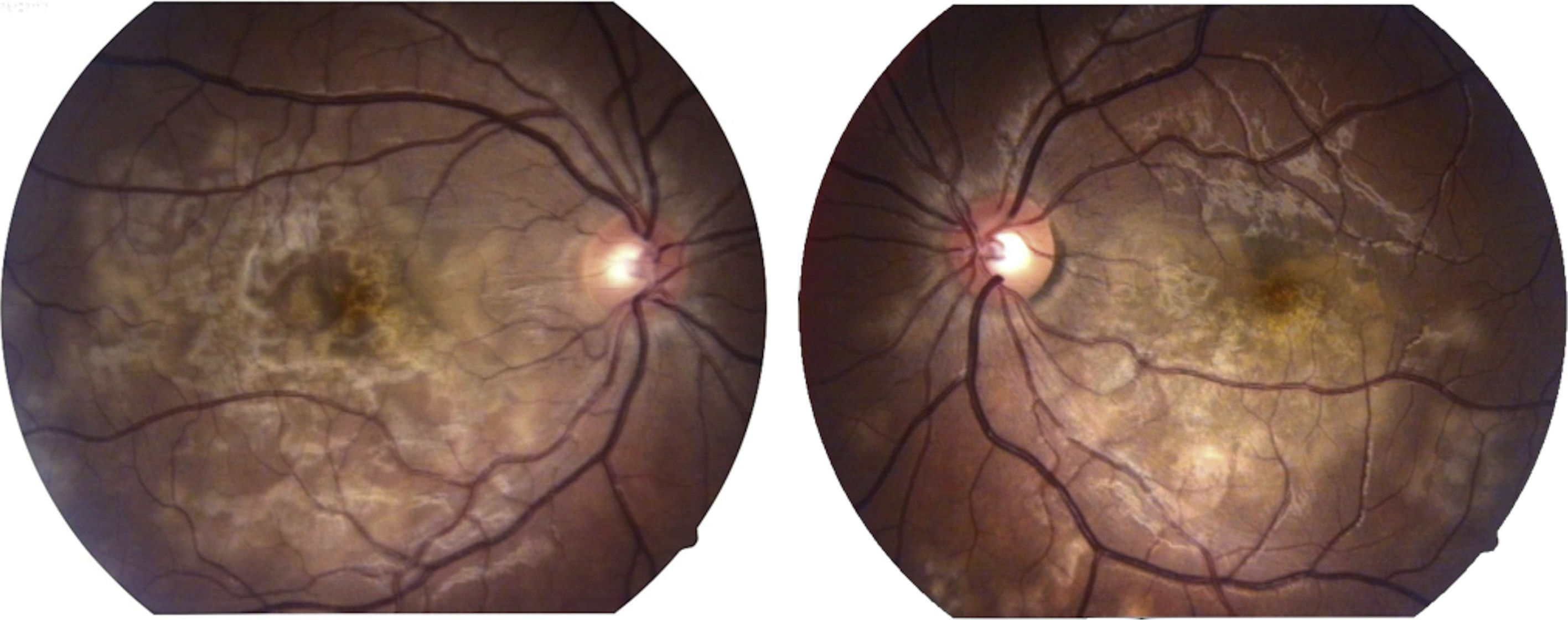
En lo que se refiere a hallazgos en la exploración del fondo de ojo, Gass1 describió la presencia de lesiones planas de color cremoso confluentes, con márgenes poco definidos dispersas en el polo posterior. Las lesiones aparecen por detrás del ecuador retiniano y son bilaterales. Se desarrollan nuevas lesiones a lo largo de varias semanas después del inicio de los síntomas, por lo que a veces aparecen lesiones visibles en diferentes etapas. Ocasionalmente se asocian con desprendimientos serosos de la retina6. Esta característica hace que sea difícil distinguir la epiteliopatía pigmentaria placoide multifocal posterior aguda de la enfermedad de Harada7,8, y algunos autores han pensado que estas entidades pueden formar parte del espectro de la misma enfermedad9.
Aunque la vitritis no es un componente significativo de la epiteliopatía pigmentaria placoide multifocal posterior aguda, la presencia y grado de inflamación intraocular varía ampliamente. Se han descrito: uveítis anterior, uveítis anterior granulomatosa; así como infiltraciones del estroma corneal10.
En general hay una mejoría de los síntomas visuales en 2 a 4 semanas, por lo que es autolimitado.
La epiteliopatía pigmentaria placoide multifocal posterior aguda tiene un pronóstico relativamente bueno en comparación con otros síndromes de puntos blancos placoideos. Sin embargo, Fiore et al.4 mostraron que aproximadamente el 50% de los pacientes tiene una recuperación incompleta de la visión, por lo que hasta el 25% de los pacientes tiene visión 20/40 o peor. El sesenta por ciento de los pacientes tiene síntomas visuales residuales.
En este sentido, la afectación foveal en el momento de presentación es un factor pronóstico no favorable, el 88% de los ojos sin afectación foveal llegan a tener una recuperación visual completa, en contraste con el 53% de los ojos que presenta afectación foveal. Gass1 informó de que la recuperación visual podría continuar durante meses después de que las lesiones se curaran, incluso hasta 6 meses después.
Las lesiones iniciales al nivel del polo posterior llegan eventualmente a desaparecer, dejando a veces algunas áreas de atrofia corioretiniana; solamente algunos casos llegan a tener un resultado visual final no favorable, como reportaron Fiore et al.4 sobre todo si esas áreas de atrofia llegan a involucrar el centro foveal.
Aunque es uno de los tipos de coriocapilaritis que se llegan a ver con relativa frecuencia, sigue siendo una entidad de la cual no se han llegado a reportar muchos casos en la bibliografía nacional e internacional.
Caso clínicoPaciente varón de 26 años de edad, que acude a la consulta refiriendo baja visual de inicio repentino de 3 días de evolución, acompañada de fotopsias en ambos ojos. El paciente no refirió antecedentes de importancia para el padecimiento actual.
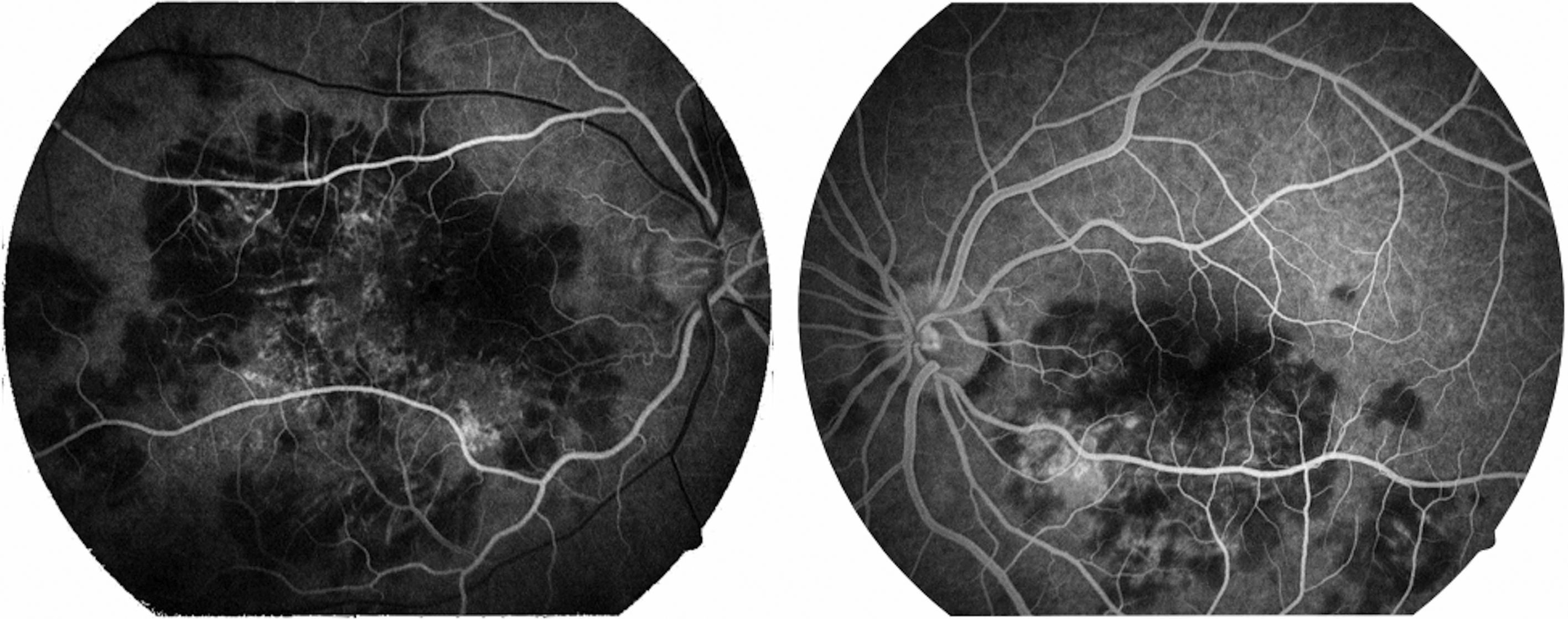
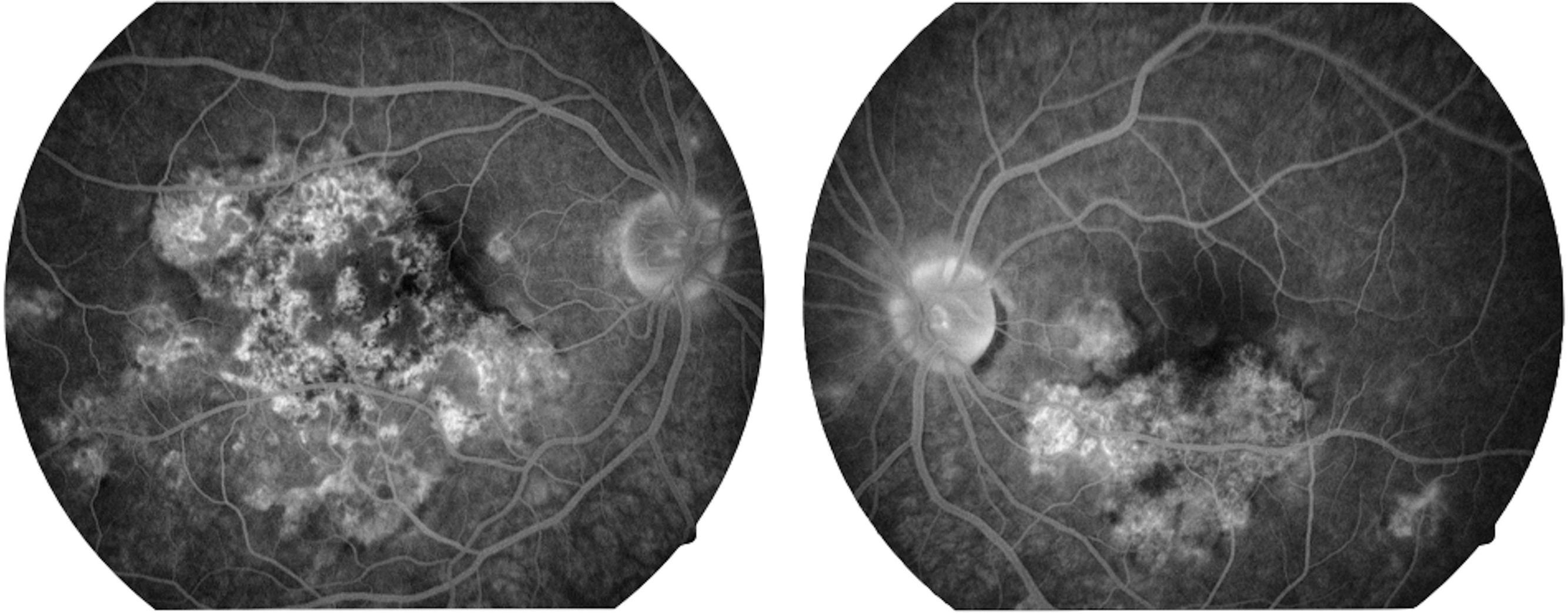
La capacidad visual fue de 20/200 en el ojo derecho y de 20/80 en el ojo izquierdo. No se encontró reacción inflamatoria en la cámara anterior ni tampoco la presencia de vitritis. Se encontraron lesiones limitadas al polo posterior de aspecto cremoso blanquecino subretinianas en forma de placas, más o menos bien definidas, con cierto grado de confluencia entre ellas (fig. 1). En la fluorangiografía retiniana se encontró hipofluorescencia en fases tempranas que se correspondía con las lesiones apreciadas en fotos clínicas en el polo posterior (fig. 2), con hiperfluorescencia de las mismas en fases tardías del estudio (fig. 3).



Imagen fluorangiográfica en fases tardías de ambos ojos. Se aprecian zonas de hiperfluorescencia en algunas de las zonas que previamente, en las fases tempranas del estudio, eran hipofluorescentes en ambos ojos. Este cambio de hipofluorescencia temprana a hiperfluorescencia tardía de las lesiones es característico de las lesiones de la epiteliopatía pigmentaria placoide multifocal posterior aguda.
En función de los datos clínicos, la imagen clínica del polo posterior de ambos ojos, así como angiográficos, se integró el diagnóstico de epiteliopatía pigmentaria placoide multifocal posterior aguda.
El cuadro evolucionó hacía la mejoría y resolución de las lesiones, con capacidad visual final de 20/40 en el ojo derecho y 20/25 en el ojo izquierdo a las 6 semanas de seguimiento sin tratamiento.
DiscusiónEl diagnóstico de epiteliopatía pigmentaria placoide multifocal posterior aguda es principalmente clínico, y se basa en el examen del polo posterior y en la evolución del cuadro a través del tiempo. En el caso del paciente, el diagnóstico se confirmó por la baja de visión, ausencia de otros antecedentes, junto con la presencia de lesiones en forma de placa que se apreciaron mediante la exploración de biomicroscopía con lente de 3 espejos de Goldman, aunado a los hallazgos de la angiografía fluoresceínica, en donde se apreció la presencia de zonas hipofluorescentes en fases tempranas del estudio con hiperfluorescencia tardía (lo cual es característico de la enfermedad) en zonas correspondientes a lesiones clínicas, aunado a lo autolimitado. Por otro lado, los síndromes de puntos blancos, así como otras entidades, se encuentran en el diagnóstico diferencial de esta enfermedad; estas incluyen: coroidopatía serpiginosa (que debe ser considerada en casos recurrentes, crónicos), coriorretinitis placoidea implacable, que debe ser tomada en cuenta en casos severos, persistentes y recurrentes (que no fue el de nuestro paciente), y el síndrome de puntos blancos evanescentes. En el diagnóstico diferencial se incluyen: neurorretinitis subaguda unilateral difusa, enfermedad de Harada, tuberculosis ocular, sarcoidosis, enfermedad fúngica, metástasis coroidea o infiltrado linfoideo, sífilis, las oclusiones vasculares de retina, la coroidopatía serosa central y el desprendimiento regmatógeno de retina.
Ante la sospecha de enfermedad de Harada debe analizarse el líquido cefalorraquídeo, para determinar la presencia pleocitosis.
El estudio que más ayuda a apoyar el diagnóstico es la fluorangiografía retiniana.
Gass1 describió las lesiones en la fase temprana como hipofluorescentes, y más adelante en el estudio hay una tinción hiperfluorescente progresiva, irregular de las lesiones (lo anterior es un dato clave presente en el estudio fluorangiográfico de nuestro paciente). A medida que el proceso se vuelve inactivo, se aprecia hiperfluorescencia correspondiente a defectos de ventana en el epitelio pigmentado de la retina, en donde el fenómeno de tinción ya no es evidente.
Otro tipo de estudios que se llegan a realizar son: la angiografía con verde de indocianina, la tomografía de coherencia óptica, en donde se han reportado anormalidades en las capas externas de la retina8. Por otro lado, la epiteliopatía pigmentaria placoide multifocal posterior aguda se ha relacionado con manifestaciones del sistema nervioso central11, que incluye: vasculitis cerebral, meningoencefalitis y enfermedad vascular cerebral.
La cefalea es un síntoma común y la epiteliopatía pigmentaria placoide multifocal posterior aguda ha llegado a imitar a la migraña incluso con aura.
A pesar de que muchos pacientes refieren algún antecedente de enfermedad viral, los síntomas de malestar general y cefalea pueden estar más relacionados con una vasculitis subyacente generalizada. Las vasculitis sistémicas12 han sido implicadas en la epiteliopatía pigmentaria placoide multifocal posterior aguda; también se ha asociado con: eritema nudoso, colitis ulcerativa, tiroiditis, nefritis, la artritis reumatoide juvenil y las enfermedades granulomatosas tales como: la granulomatosis de Wegener (granulomatosis con poliangeítis) y la tuberculosis pulmonar.
En la actualidad algunos creen que aparece una lesión vascular que afecta a la coroides, que puede causar isquemia coroidea parcial que conduce a daños en el epitelio pigmentado de la retina, y en última instancia afectar a los fotorreceptores. Los estudios de imagen son compatibles con esta aseveración. Es posible que un proceso primario que implica la retina externa y el epitelio pigmentado de la retina podría secundariamente causar anormalidades de la coroides en la angiografía.
Parece que puede haber un efecto gatillo, ya sea de naturaleza inflamatoria o infecciosa que incita a este proceso. Además, asociaciones con HLA-B7 y HLA-DR2 han sido descritas, lo que sugiere que ciertos individuos son más susceptibles. Aunque la etiología exacta es desconocida, puede haber asociación con la enfermedad viral como los adenovirus13. En la mitad de los casos presentados por Ryan y Maumenee2 hubo manifestaciones prodrómicas de enfermedad viral, y también se ha descrito la asociación con infecciones bacterianas; en este proceso hay una activación de los linfocitos T sensibilizados por diversos estímulos tales como: bacterias, virus y hongos. Las células T activadas liberan linfocinas, que a su vez activan macrófagos y células T citotóxicas. Los macrófagos, a continuación, dan lugar a células epitelioides y células gigantes que pueden conducir a la formación de granulomas.
Las asociaciones sistémicas, tales como la inflamación del sistema nervioso central que se mencionó anteriormente, podrían explicarse de esta manera. La presencia de un proceso trombótico de la coroides también se ha llegado a postular.
No existe tratamiento específico, incluidos los esteroides intravenosos en pacientes afectados del sistema nervioso central, también se usan inmunosupresores14. Nuestro paciente solo se vigiló.
Rara vez se desarrollan neovascularizaciones coroideas en las que los antiangiogénicos intravítreos tienen utilidad.
ConclusionesLa epiteliopatía pigmentaria placoide multifocal posterior aguda es una entidad inflamatoria, por lo general benigna, poco común, de carácter autolimitado que afecta predominantemente a capas externas de la retina, así como a la coriocapilaris. Ocasionalmente llega a involucrar al sistema nervioso central, y debe formar parte del diagnóstico diferencial de los procesos inflamatorios que afectan el polo posterior, principalmente de los llamados síndromes de puntos blancos.
Es importante no dar terapéuticas innecesarias, que lejos de ayudar al paciente pueden llegar a ocasionarle mayor perjuicio.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
El autor agradece la participación del personal de la Clínica David en la realización de este reporte de caso.