


Combined hepatocellular-cholangiocarcinoma is a rare primary hepatic tumour, showing both hepatocellular as well as biliary epithelium differentiation. Its diagnosis is often delayed, as it occurs in young patients without comorbidities and with non-specific symptoms. Most cases are confused with other types of cancer, especially fibrolamellar liver cancer, which is more frequent and has similar clinical and radiological features.
Clinical caseThe case is presented of a 26 year old woman with a giant combined hepatocellular-cholangiocarcinoma with difficulties in its diagnosis and a complicated surgical approach.
DiscussionThe definitive diagnosis of this disease is defined by the histological demonstration of cholangiolar and hepatocellular differentiation, with surgical treatment always being the best choice, but with lower survival than classic hepatocellular carcinoma and cholangiocarcinoma. In some patients with unfavourable prognostic factors, adjuvant chemotherapy mainly directed cholangiolar component can be given.
ConclusionThe current incidence of combined hepatocellular-cholangiocarcinoma varies from 2% to 5% of cases, and is one of the rarest histological types in the world. The large size and hypervascularisation of the tumour makes a surgical approach difficult in these patients, while the rare histological features require a more detailed study of the piece and the application of immunohistochemical techniques to confirm the diagnosis.
El hepatocolangiocarcinoma es una entidad poco frecuente de tumor primitivo hepático que muestra diferenciación tanto hepatocelular como del epitelio biliar. Su diagnóstico suele ser tardío, ya que se presenta en pacientes jóvenes, sin enfermedades asociadas y con síntomas inespecíficos. La mayoría de los casos se confunde con otro tipo de carcinoma, sobre todo con el hepatocarcinoma fibrolamelar, por ser más frecuente y presentar características clínicas y radiológicas similares.
Caso clínicoPresentamos el caso de una paciente de 26 años con un hepatocolangiocarcinoma gigante, con dificultades en su diagnóstico y un complicado abordaje quirúrgico.
DiscusiónEl diagnóstico definitivo de esta enfermedad se define por la demostración histológica de una diferenciación dual hepatocelular y colangiolar. El tratamiento quirúrgico es siempre de elección, con una supervivencia inferior a la del hepatocarcinoma y de los colangiocarcinomas clásicos. En algunos pacientes con factores pronósticos desfavorables se puede dar adyuvancia con quimioterapia, dirigida principalmente al componente colangiolar.
ConclusiónLa incidencia real del hepatocolangiocarcinoma varía entre el 2 y el 5% de los casos, y es uno de los tipos histológicos más raros que existen. El gran tamaño y la hipervascularización de la tumoración dificultan el abordaje quirúrgico en este tipo de pacientes, mientras que las características histológicas, poco frecuentes, requieren un estudio más detallado de la pieza y la aplicación de técnicas inmunohistoquímicas que confirmen el diagnóstico.
Hepatocholangiocarcinoma is a rare primary hepatic tumour showing both hepatocellular as well as biliary epithelium differentiation.1 Its diagnosis is often delayed, as it occurs in young patients without comorbidities and with non-specific symptoms.1,2 Most cases are confused with other types of cancer,3 especially fibrolamellar liver cancer, which is more frequent and has similar clinical and radiological features.4
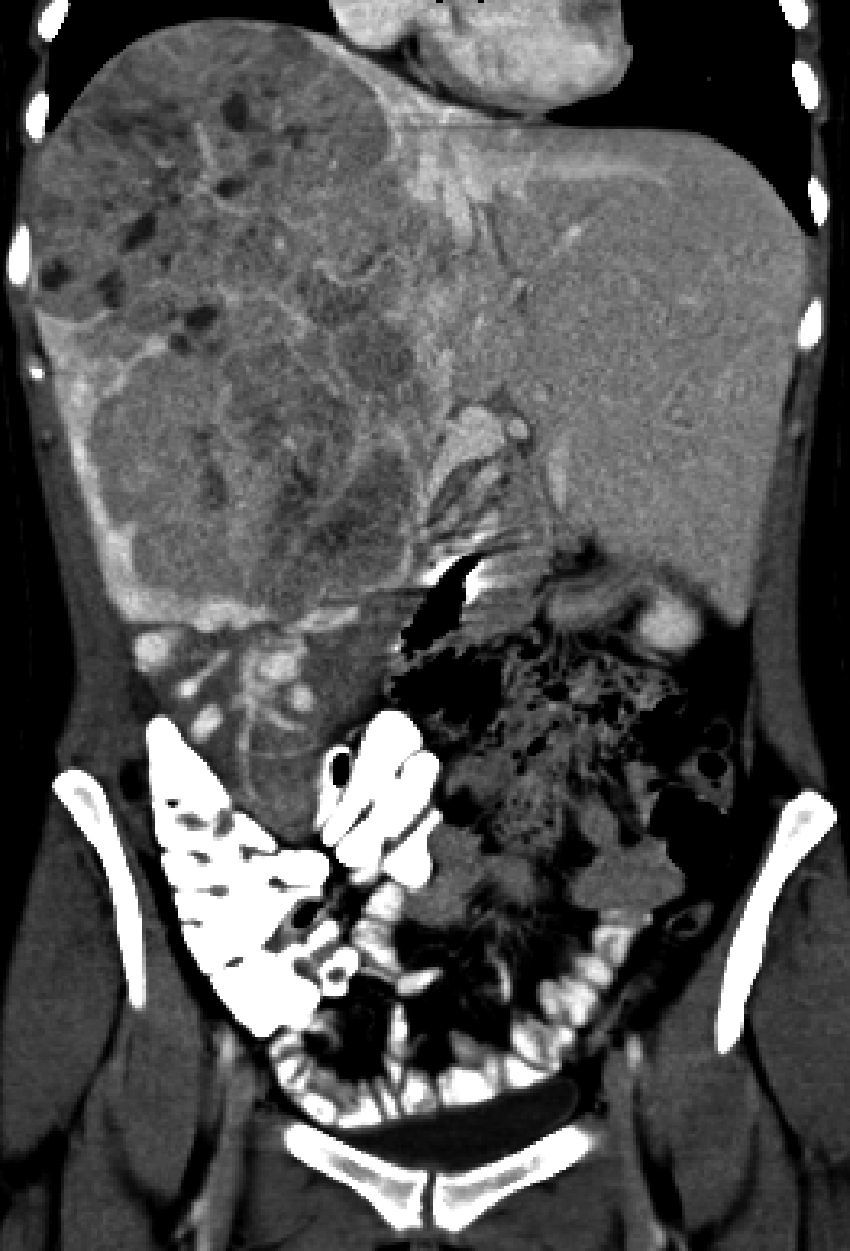
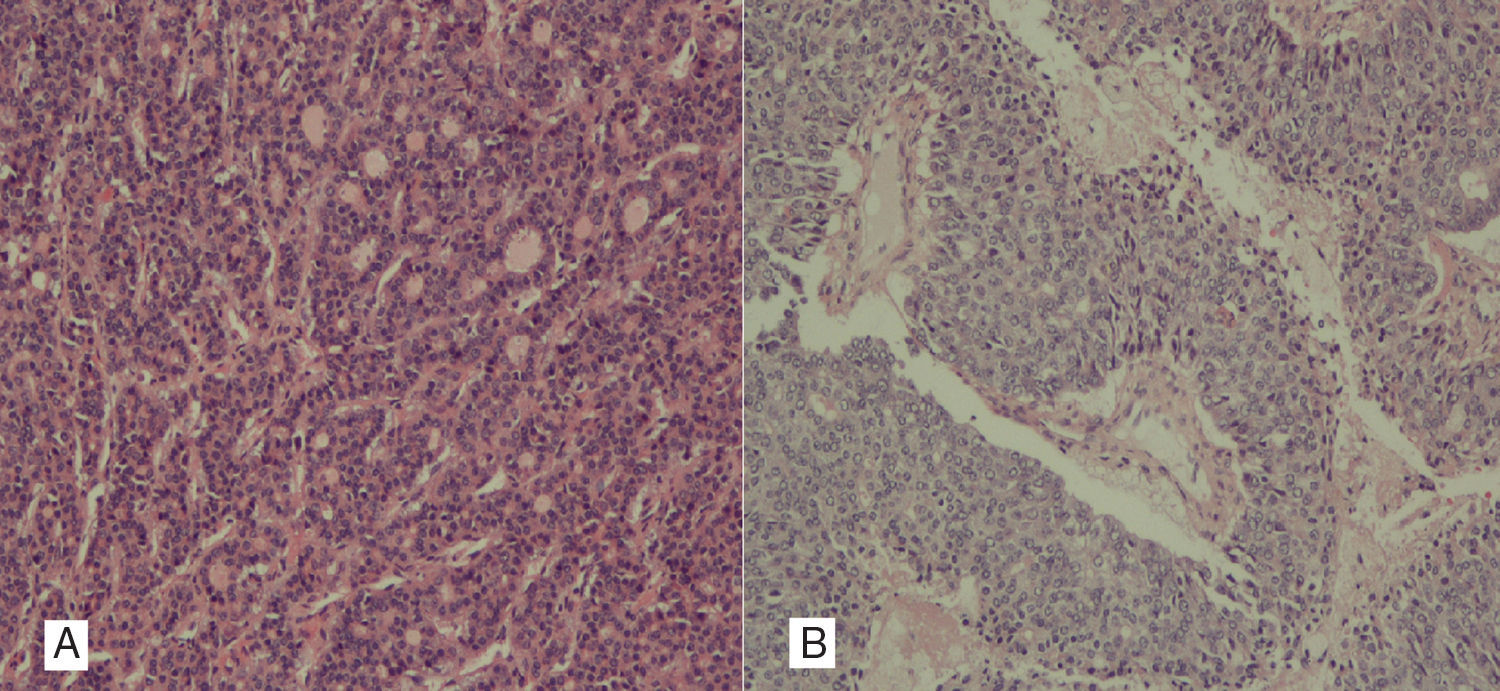
Clinical caseA 26 year old female patient with no medical history of interest, presented with abdominal pain and diarrhoea of one year duration, with a palpable mass on physical examination. Abdominal and pelvic computed axial tomography was performed and a hepatic mass measuring 20×20×14cm occupying the right lobe (segments VIII, VII) and partially segment VI was observed. Lobe contours were well defined, with arterial enhancement and lavage in the portal phase and hyperdense focal areas due to necrosis. Radiological findings observed were also suggestive of fibrolamellar liver cancer (Fig. 1). The tumour was in close contact with the left suprahepatic vein, probably invading segment IVa. There was extremely highly marked collateral venous circulation flowing through segments VI and V, which drained into the left and right hepatic veins. A surgical approach was taken and a giant liver tumour was observed. It was attached and had replaced the entire right liver lobe, measured 25×18cm, stretching further than the line of the round ligament, up to segment II and occupying practically the whole of segment IV, with hypervascularisation in all regions. Intraoperative ultrasound ruled out further satellite lesions. Anterior approach for liver transection was performed with some difficulty, due to extended neoformation vascularisation which led to severe bleeding, required 6 clamps, and a total time in surgery of 1h 47min. Right trisectionectomy, with minor extension to segment II was performed. The patient's evolution was favourable with moderate anaemisation which did not require any transfusions, and with no complications showing up in X-rays. Post procedure ascitic fluid continued to be present in the surgical site and was controlled with diuretics. The patient was discharged one week after surgery. At present, 7 months after surgery, the patient is asymptomatic and there is no sign of tumour activity. The final histological study shows the tumour measuring 14.7×9cm with free margins and a mixed hepato-biliary cancer or combined hepatocholangiocarcinoma, formed by the 2 types of tumours, hepatocarcinoma and cholangiocarcinoma, in clearly distinguishable areas, and no transition area. This was a mixed hepatocholangiocarcinoma, of the classic WHO type with 2 morphological and immunohistochemical patterns. The hepatocarcinoma is made up of trabecular and glandular tumours, whilst the cholangiocarcinoma is a desmoplasic, tubuloglandular, more featureless tumour. Furthermore, the immunohistochemical techniques practised show that several regions of the tumour tested positive for Hepatocyte paraffin 1 (Hep-Par1), the tumour marker corresponding to the hepatocarcinoma type component (Fig. 2A) and in others, tested positive for cytokeratin AE1-AE3, 7 and villin, standard findings of cholangiocarcinomas (Fig. 2B).
 and partially segment VI with well defined lobulated contours, arterial enhancement and portal-phase lavage. Hypodense focal regions persist due to necrosis. Radiologic findings suggestive of fibrolamellar liver cancer.")
Computed axial tomography of the abdomen and pelvis in arterial phase, liver mass of 20×20×14cm occupying the right lobe (segments VIII, VII) and partially segment VI with well defined lobulated contours, arterial enhancement and portal-phase lavage. Hypodense focal regions persist due to necrosis. Radiologic findings suggestive of fibrolamellar liver cancer.
 Histological image with pseudoglandular pattern of hepatocarcinoma, with formation of glandular lumen. Positive Hepatocyte paraffin 1 (Hep-Par1), negative cytokeratins 7 and 20. (B) Non-differentiated cholangiocarcinoma, in which glandular lumen are distinguished and areas of intratumoral necrosis. AE1-AE3 and 7 cytokeratins tested positive; Hep-Par1 and cytokeratin 20 tested negative.")
(A) Histological image with pseudoglandular pattern of hepatocarcinoma, with formation of glandular lumen. Positive Hepatocyte paraffin 1 (Hep-Par1), negative cytokeratins 7 and 20. (B) Non-differentiated cholangiocarcinoma, in which glandular lumen are distinguished and areas of intratumoral necrosis. AE1-AE3 and 7 cytokeratins tested positive; Hep-Par1 and cytokeratin 20 tested negative.
The real incidence of hepatocholangiocarcinoma is unknown but varies between 2% and 5%1–3; it is one of the rarest histological types in existence. Its histogenesis is unclear, but it probably originates from intermediate type cells or progenitor dual type cells, with no relation to the presence of liver cirrhosis, unlike hepatocarcinoma.2 There are risk factors which contribute to the development of this cancer, such as hepatitis B virus infection, excessive consumption of alcohol, a family background of liver cancer and diabetes mellitus, but there is no relationship with hepatitis C virus infection.5
Clinical symptoms are usually the same as those of hepatocarcinoma where differential diagnosis is required with other tumours which attach to healthy liver tissue or with benign liver diseases such as focal nodular hyperplasia.6 Final diagnosis is defined by histological demonstration of a dual hepatocellular and cholangiolar differentiation, which may be of 3 types: double, when there is the presence of hepatocarcinoma and cholangiocarcinoma in the same liver but they are separate; combined, when both carcinomas are contiguous and mingle on growth; or mixed, when a single tumour mass which is closely intermingled is present.3,7 Recent WHO classification unites the previous ones into one classic type group and differentiates it from another group divided according to the presence of intermediate cells with different immune-phenotype stem cells.8 The histological degree of malignancy is divided using a scale from I to IV in accordance with the increase of nuclear irregularities, hyperchromatism and nuclear/cytoplasm ratio.4 In our case this would be classified as a classic type by the WHO (standard areas of hepatocarcinoma and cholangiocarcinoma)8 and an Allen and Lisa type B (combined or collision type which are contiguous because they mingle when they grow, but there are still boundaries between the two populations).7 Immunohistochemical study may clarify doubts regarding diagnosis and confirm the cholangiolar carcinoma component with positive testing of the carcinoembryonic antigen, cytokeratin 7 and CA19-9, and that of hepatocarcinoma with positive testing for Hep-Par1 and alpha-fetoprotein. Both should be present in at least 10% of the tumour for consideration as hepatocholangiocarcinoma.2,3
ConclusionSurgical treatment is always of choice for hepatocholangiocarcinoma, with a lower survival rate to classic hepatocarcinoma and cholangiocarcinomas.2,9 In some patients with poor prognostic factors, adjuvant treatment with chemotherapy, mainly targeted at the cholangiolar component may be given. This component is the one most related to relapse and the presence of distant disease.10 The large size and hyper vascularisation of the tumour hinders a surgical approach in this type of patient and currently this is the only effective treatment, whilst the infrequent histological characteristics require more detailed study of the specimen and the application of immunohistochemical techniques to confirm diagnosis.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of interestsThe authors have no conflict of interest to declare.
Please cite this article as: Tejera-Hernández AA, Cabrera-García ME, Martínez-Martin MS, García-Plaza G, Larrea-Olea FJ, Hernández-Hernández JR. Hepatocolangiocarcinoma en paciente joven con tumoración hepática gigante. Cir Cir. 2017;85:250–253.