



Androgen insensitivity syndrome is an X-linked disorder, and is characterised by a female phenotype in a chromosomally male individual. It usually occurs in puberty with primary amenorrhoea or as an inguinal tumour in a female infant. In recent years, it is often also diagnosed in fertility clinics in adulthood.
ObjectiveThe case is presented of a pure seminoma in a woman with the reference diagnosis of inguinal hernia.
Clinical caseA 53 year old woman, who was operated on in 2014 due to a nodule in left groin. Androgen insensitivity syndrome was corroborated, and histopathology reported it as a right testicular seminoma.
DiscussionThe importance of early diagnosis is discussed, highlighting the consequences of misdiagnosis, and question whether these patients have been adequately treated in the past. The risk of malignant transformation of an undescended testicle increases with age, thus gonadectomy should be performed after puberty, and in some cases hormone replacement therapy.
El síndrome de insensibilidad a los andrógenos es un trastorno ligado al cromosoma X que se caracteriza por un fenotipo femenino, en un individuo cromosómicamente masculino. Por lo general, se presenta en la pubertad con amenorrea primaria o como un tumor inguinal en un lactante de sexo femenino. En los últimos años, también se suele diagnosticar en clínicas de fertilidad en la edad adulta.
ObjetivoPresentamos un caso de seminoma puro, en una mujer con el diagnóstico de referencia de hernia inguinal.
Caso clínicoMujer de 53 años, la cual se operó en el año 2014 por un nódulo en la ingle izquierda. Se corroboró síndrome de insensibilidad a los andrógenos, y en la revisión histopatológica se reportó como un seminoma de testículo derecho.
DiscusiónSe discute la importancia del diagnóstico precoz, destacamos las consecuencias de un mal diagnóstico y plantemos la cuestión de si estos pacientes han sido tratados adecuadamente en el pasado. El riesgo de transformación maligna de testículo no descendido aumenta con la edad, por lo que la gonadectomía se debe realizar después de la pubertad y en algunos casos, añadir terapia de reemplazo hormonal.
Androgen insensitivity syndrome is an X-linked disorder, with an incidence of between 1:20,000 and 1:64,200 live births. An affected patient is genetically male, with a 46 XY Karyotype and with normal differentiation of the testicles, with no uterus. It is the third most common cause of primary amenorrhea and is the most common form of male pseudohermaphrodite.1,2
This condition was first described by Morrisen3 in 1953, from 80 cases collected from the literature and 2 cases of his own and it was called “testicular feminisation syndrome”. The said patients presented a female phenotype, little body hair, normal external genitals of a female and the presence of testicles. A short while later, together with Mahesh,4 cases were described which differed from those initially described and they were considered to correspond to an incomplete form. In 1947, Reifenstein5 described a syndrome characterised by hypospadias, gynecomastia and infertility, with an increase in the follicle-stimulating hormone, linked to chromosome X. Thanks to the works by Keenan6 it could be demonstrated that this syndrome was characterised by the lack of response of the peripheral tissues to the action of testosterone and that the locus of this condition was to be found in chromosome Xq11-12.7 A study on the Mexican population reported a rate between 3.6% at 25 years of age to 33% at 50.8 We present the case of a woman with an inguinal tumour which turned out to be an undescended testicle with a germinal neoplasm (Table 1).
Classification of phenotype severity of AIS.
| Level | Definition |
|---|---|
| 1 | Masculine phenotype, infertility by azoospermia (resistance to minimal androgen levels or Kennedy syndrome) |
| 2 | Partial AIS with masculine phenotype: hypospadias |
| 3 | Partial AIS with masculine phenotype, small penis, perineoscrotal hypospadias, bifid scrotum |
| 4 | Partial AIS with ambiguous phenotype, penis similar to phallus, labioscrotal folds and single perineal opening |
| 5 | Partial AIS with female phenotype: separate urethral and vaginal orifices, minimal foetal androgenous action, clitoromegaly |
| 6a | Partial AIS with female phenotype, female type genitals, no foetal androgenisation. Androgenic development in puberty. |
| 7a | Complete AIS with female phenotype and absence of pubic and underarm hair after puberty |
AIS: androgen insensitivity syndrome.
Levels 6 and 7 of sexual ambiguity, proposed by Quigley9 are not distinguished in the prepubertal stage.
A female aged 53 who stated her development at puberty had been subject to irregular menstrual cycles, who had not had any abortions and who was experiencing infertility. She had a surgical history of excision of a left inguinal tumour. She was referred to our hospital with histopathology report of dysgerminoma type left ovarian germ-cell tumour.
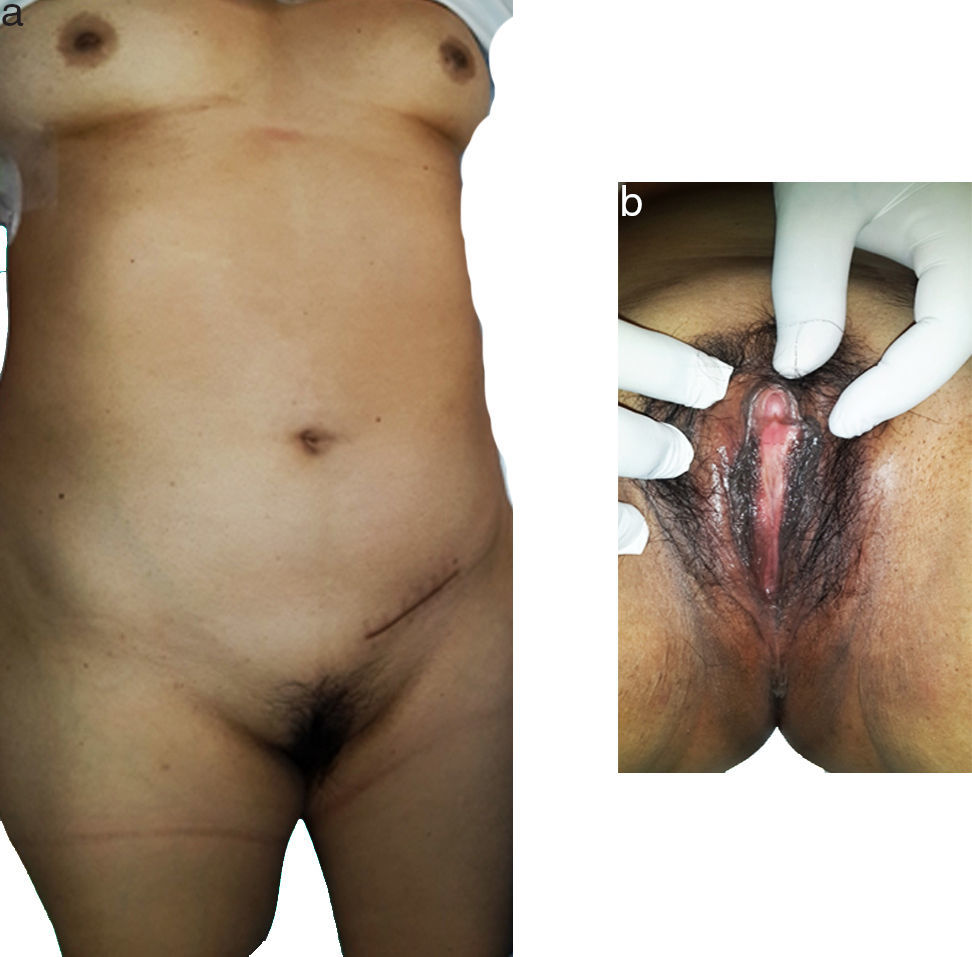
Physical examination did not reveal any pathological facies; there was presence of female type mammary glands and normal female external genitals, not much underarm hair and a palpable tumour in the right groin area (Fig. 1). On digital vaginal examination there was an absence of any cervix and no uterus.
 Mammary development is observed, gynecoid hair and the surgical scar from previous surgery in the left inguinal region. (b) External female genitals are observed.")
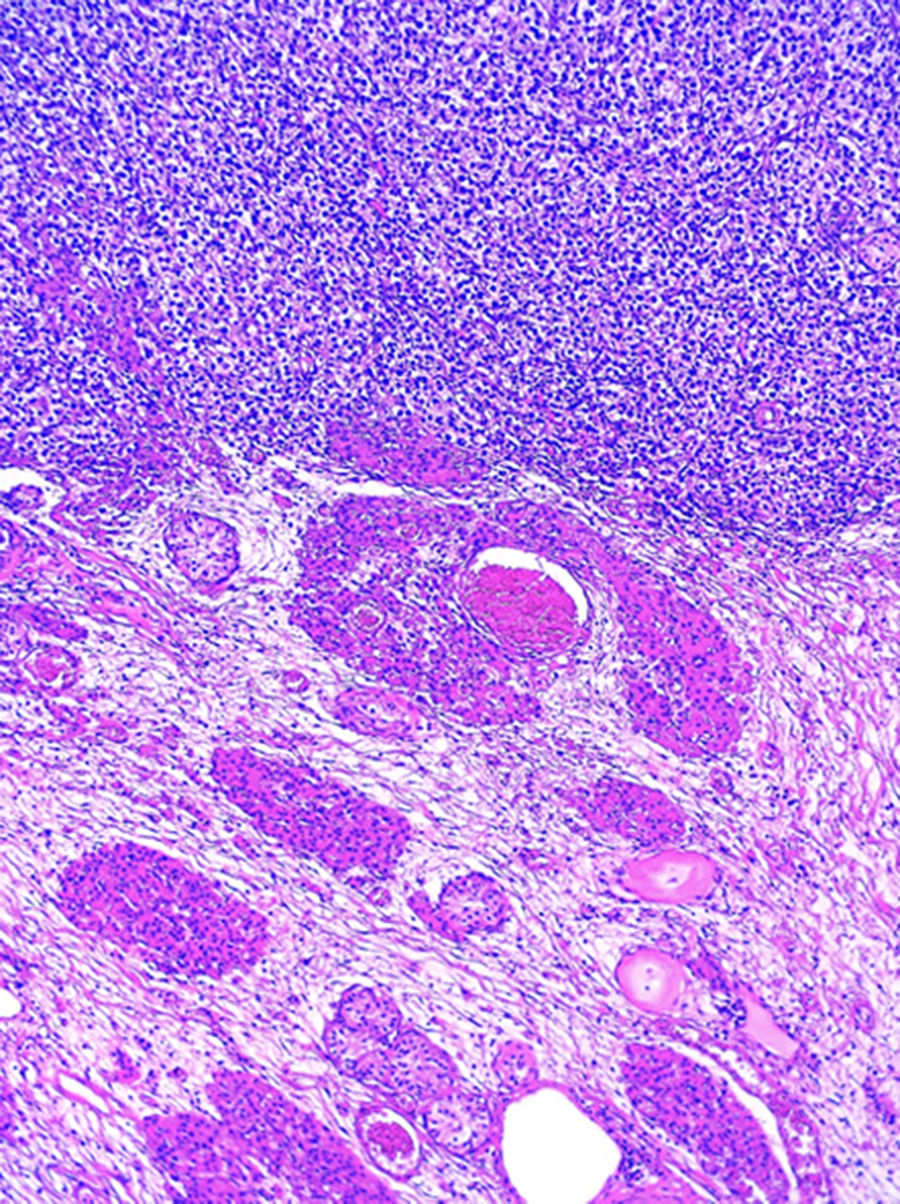
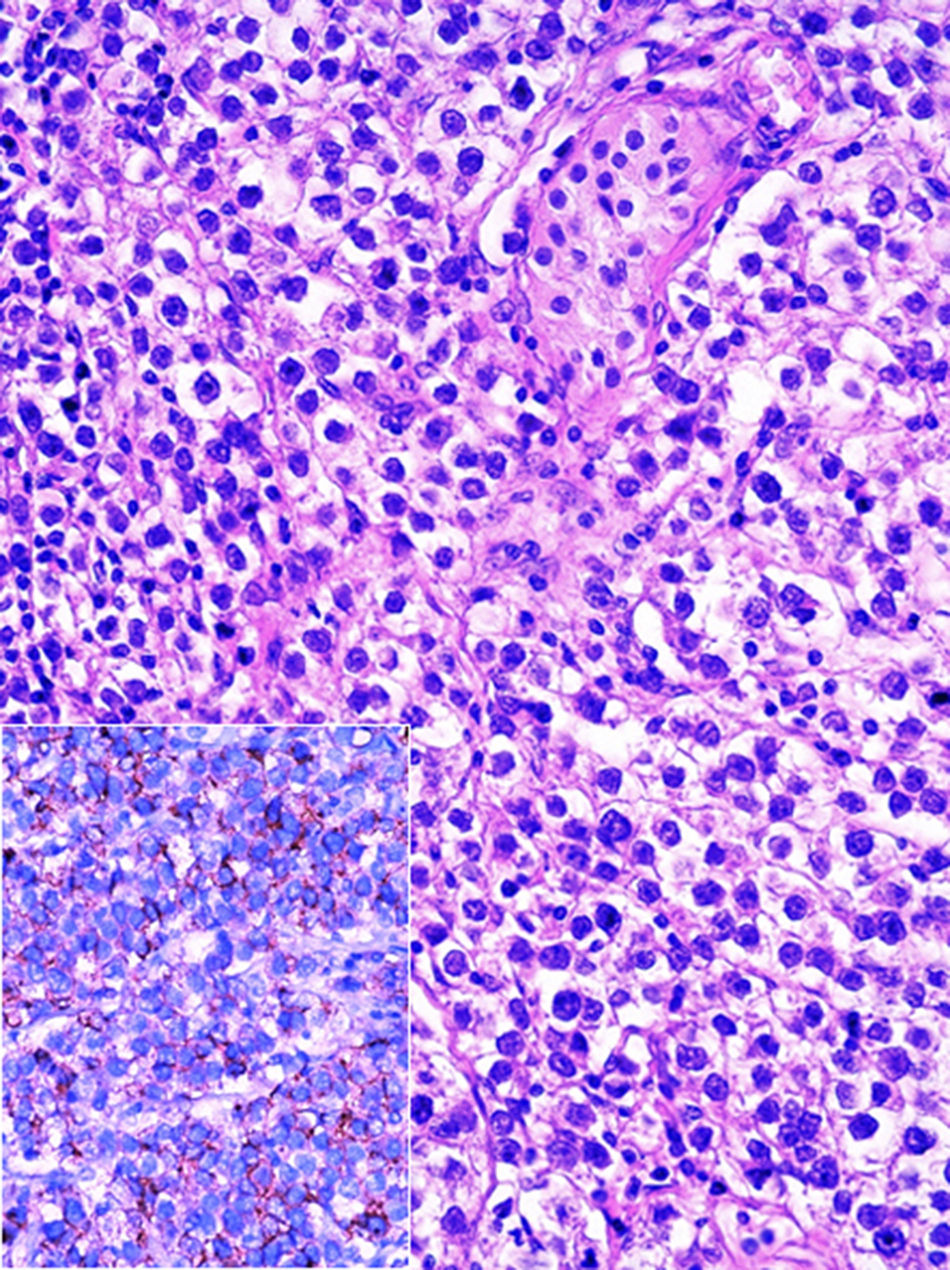
Histopathological examination of blocks and slides referred to as “left ovary” revealed testicular parenchyma with atrophy and pure classical seminoma type germ-cell tumour (Fig. 2). In the residual testicle intratubular focal germinal neoplasm was identified.
, with the presence of classical pure seminoma (upper portion of the photograph) (haematoxylin and eosin, X100).")
With regard to the patient's hormonal status, it was similar to that described by other authors, with a serum testosterone level prior to surgery of 38,900ng/dL (normal masculine is from 240–950ng/dL), a luteinizing hormone level of 31,531.50mU/mL (normal masculine is from 1 to 10mU/mL), human chorionic gonadotropin of 10.60U/mL (normal 5U/mL), alpha-fetoprotein of 2.77ng/mL (normal 15ng/mL), carcinoembryonic antigen of 1.19ng/mL, CA-125 of 7.49U/ml, and LDH of 213UI/l (normal range from 114 to 198UI/L).
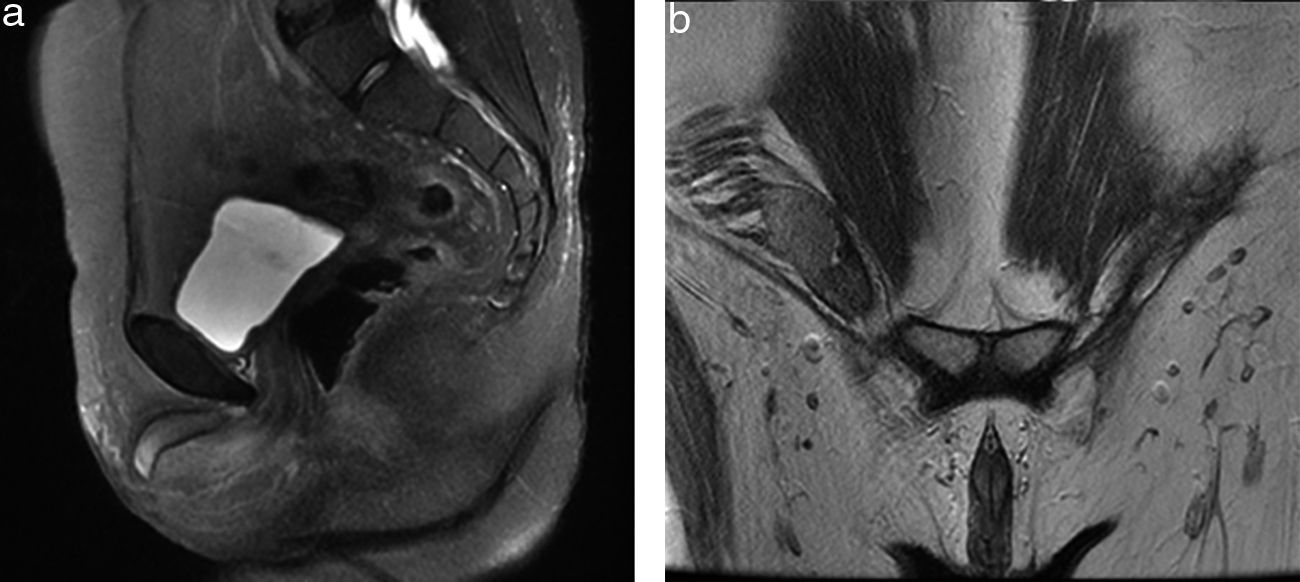
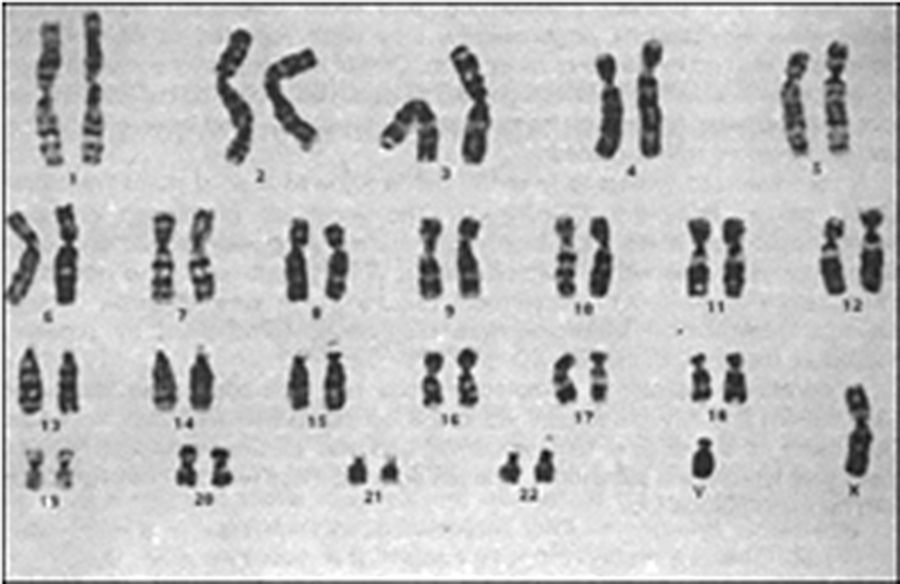
Contrast-enhanced computed tomography was performed, in coronal and axial plane with observation of a retroperitoneal ganglionic conglomerate of paraaortic location. This extended immediately below the renal vein, the right cryptochidism through the internal inguinal ring and post-surgery changes in the left inguinal region (Fig. 3). Chromosomic and karyotype analysis showed the presence of 46XY (Fig. 4).
 in sagittal plane; (b) in coronal plane, where the absence of the uterus and both ovaries is observed, and the presence of the right testicle within the ipsilateral inguinal canal is observed, together with the presence of corpora cavernosa and trace of genital tubercle.")
Magnetic resonance of the pelvis. Sequence enhanced in T2: (a) in sagittal plane; (b) in coronal plane, where the absence of the uterus and both ovaries is observed, and the presence of the right testicle within the ipsilateral inguinal canal is observed, together with the presence of corpora cavernosa and trace of genital tubercle.

At an interdisciplinary session it was decided to use surgery. The product from the right orchiectomy measured 4.6×2.2×2cm, and consisted of a testicle, epididymis and spermatic cord. Histopathology reported it as testicular parenchyma with atrophy, hyperplasia of Leydig cells and microcalcifications (chryptorchid testicle), and germ cell intratubular neoplasia foci, which showed expression of PLAP (Fig. 5) and C-kit. The tunica vaginalis and the spermatic cord were free from neoplasm.
 and in PLAP insert (express placental alkaline phosphatase, immunohistochemical reaction 400×).")
The patient's postoperative evolution and follow-up have been favourable, with no recurrence of the disease 12 months after surgery.
DiscussionWe present the case of a patient with karyotype 46XY, which corresponds to a confirmed case of androgen insensitivity syndrome, and who developed neoplasms in both undescended testicles, a classical pure seminoma in one testicle and in the contralateral one, the precursor lesion: a contralateral intratubular germinal neoplasm.
Androgen insensitivity syndrome is a condition which classically presents in adolescence as primary amenorrhea or in infancy as an inguinal mass, which is often the result of an undescended testicle.10,11 Treatment is bilateral orchiectomy followed by hormonal therapy, because at a later age it is associated with a higher risk of testicular malignancy.12 Psychological support and guidance are also essential, both for the patients and their families. The criteria described by Quigley9 are used to assess the level of sexual ambiguity. Over 800 mutations in the receptor antigen gene have been reported in patients with androgen insensitivity syndrome, which are spread throughout the gene with a preponderance located in the ligand binding domain. The most severe mutations are generally associated with a phenotype insensitivity syndrome to the complete androgens, but the correlation is less defined in the partial androgen insensitivity syndrome.
Androgen insensitivity syndrome is an entity with a clinical spectrum which ranges from female phenotype, as is the case we present, to mild forms of sexual ambiguity which merit genetic, hormonal and diagnostic study by imaging. This should be beneficial to the patients and to their family members, since this entity is a hereditary condition of the receptivity of androgens. Its treatment must be approached by a multidisciplinary team, with assessment of genital function and sexual identity for treatment. The opinion of the family and patient are critical here. Through this case, we stress the association of the androgen insensitivity syndrome to androgens and an inguinal tumour which highlights the risk of testicular malignancy. The 2 most common types of testicular tumours related to androgen insensitivity syndrome are those of germ cells and those of Sertoli cells. The primary risk factor for the development of these tumours is the absence of testicular descent, which represents 10% of patients with testicular tumours.13
The androgen insensitivity syndrome rate in women with a hernia diagnosis is 1.6%.14 Several retrospective studies have estimated that between 0.8% and 2.4% of girls with inguinal hernias have androgen insensitivity syndrome.15 Family cases have been published where several family members have minimal defects of virilisation (micro phallus or bifid scrotum) and more severe anomalies such as: perineoescrotal hipospadies, absence of vasa deferens and vaginal opening.16,17 One study of 150 cases18 identified mutations in 39% of patients and the establishment of the cause of pathogenesis in 60% of them, with late presentation in the majority, and little awareness of those affected and their family members.
The diagnosis of androgen insensitivity syndrome was delayed in this patient. Physicians should be aware that a careful physical examination and detailed gynaecological examination in all patients with primary amenorrhea, infertility and inguinal tumour is required. In the past we believe many cases of inguinal masses were considered to be ectopic tissue, fibroids, leiomyomas or benign neoplasias of mesenchymal origin, but it is clear that the patients should know if the procedure was a direct repair of the hernia or whether a gonad was found and therefore whether it was eliminated. In our case, it appears that the history of left inguinal surgery led to an erroneous assumption that an inguinal hernia plasty was performed. This demonstrates the serious consequences of making assumptions with regard to patient management with a vague medical background, which does not clearly show that orchiectomy had been performed. Histopathology review confirmed that the neoplasm of tissues extracted from the inguinal canal was a left testicle and the right one was observed in computed axial tomography and later removed. The precursor lesion of a germ cell tumour was discovered, the germ cell intratubular neoplasm, which if had not been diagnosed and removed, would probably have developed into another tumour.
A 5 year survival rate for low grade testicular tumour is 90–95%,13 and life expectancy and prognosis of this patient is therefore favourable.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Herrera-Gómez A, García-Pérez L, Gallardo-Alvarado L, Isla-Ortiz D, Salcedo-Hernández RA, Chanona-Vilchis J. Mujer con seminoma puro y neoplasia intratubular germinal contralateral. Informe de un caso. Cir Cir. 2017;85:245–249.