



El síndrome de insensibilidad a los andrógenos es un trastorno ligado al cromosoma X que se caracteriza por un fenotipo femenino, en un individuo cromosómicamente masculino. Por lo general, se presenta en la pubertad con amenorrea primaria o como un tumor inguinal en un lactante de sexo femenino. En los últimos años, también se suele diagnosticar en clínicas de fertilidad en la edad adulta.
ObjetivoPresentamos un caso de seminoma puro, en una mujer con el diagnóstico de referencia de hernia inguinal.
Caso clínicoMujer de 53 años, la cual se operó en el año 2014 por un nódulo en la ingle izquierda. Se corroboró síndrome de insensibilidad a los andrógenos, y en la revisión histopatológica se reportó como un seminoma de testículo derecho.
DiscusiónSe discute la importancia del diagnóstico precoz, destacamos las consecuencias de un mal diagnóstico y plantemos la cuestión de si estos pacientes han sido tratados adecuadamente en el pasado. El riesgo de transformación maligna de testículo no descendido aumenta con la edad, por lo que la gonadectomía se debe realizar después de la pubertad y en algunos casos, añadir terapia de reemplazo hormonal.
Androgen insensitivity syndrome is an X-linked disorder, and is characterised by a female phenotype in a chromosomally male individual. It usually occurs in puberty with primary amenorrhoea or as an inguinal tumour in a female infant. In recent years, it is often also diagnosed in fertility clinics in adulthood.
ObjectiveThe case is presented of a pure seminoma in a woman with the reference diagnosis of inguinal hernia.
Clinical caseA 53 year old woman, who was operated on in 2014 due to a nodule in left groin. Androgen insensitivity syndrome was corroborated, and histopathology reported it as a right testicular seminoma.
DiscussionThe importance of early diagnosis is discussed, highlighting the consequences of misdiagnosis, and question whether these patients have been adequately treated in the past. The risk of malignant transformation of an undescended testicle increases with age, thus gonadectomy should be performed after puberty, and in some cases hormone replacement therapy.
El síndrome de insensibilidad a los andrógenos es una condición recesiva ligada al cromosoma X, con una incidencia de entre 1:20,000 y 1:64,200 nacidos vivos. Un paciente afectado es genéticamente masculino, con un cariotipo 46XY y con diferenciación normal de los testículos, sin útero. Es la tercera causa más frecuente de amenorrea primaria y es la forma más común de pseudohermafroditismo masculino1,2.
Fue descrito por primera vez por Morrisen3 en 1953, a partir de 80 casos recolectados de la literatura y 2 casos propios y lo denominó «síndrome de feminización testicular». Dichos pacientes presentaban fenotipo femenino, escaso vello corporal, genitales externos normales de mujer y presencia de testículos. Poco después, junto con Mahesh4, describen casos que diferían de aquellos descritos inicialmente y consideraron que correspondían a una forma incompleta. En 1947, Reifenstein5 describió un síndrome caracterizado por hipospadias, ginecomastia e infertilidad, con incremento de la hormona folículo estimulante, el cual se encontraba ligado al cromosoma X. Gracias a los trabajos de Keenan6 se pudo demostrar que este síndrome se caracterizaba por falta de respuesta de los tejidos periféricos a la acción de la testosterona y que el locus de este trastorno se encontraba localizado en el cromosoma Xq11-127. Un estudio en población mexicana reporta que va desde el 3.6% a los 25 años hasta el 33% a los 50 años8. Presentamos el caso de una mujer con un tumor inguinal que resultó ser un testículo no descendido con una neoplasia germinal (tabla 1).
Clasificación de la severidad fenotípica de SIA
| Grado | Definición |
|---|---|
| 1 | Fenotipo masculino, infertilidad por azoospermia (resistencia a andrógenos mínima o síndrome de Kennedy) |
| 2 | SIA parcial con fenotipo masculino: hipospadias |
| 3 | SIA parcial con fenotipo masculino, pene pequeño, hipospadias perineoescrotal, escroto bífido |
| 4 | SIA parcial con fenotipo ambiguo, pene similar a falo, pliegues labioescrotales y orificio perineal único |
| 5 | SIA parcial con fenotipo femenino: orificio uretral y vaginal separados, acción androgénica fetal mínima, clitoromegalia |
| 6a | SIA parcial con fenotipo femenino, genitales de tipo femenino, no androgenización fetal. Desarrollo androgénico en la pubertad |
| 7a | SIA completo con fenotipo femenino y ausencia de vello púbico y axilar después de la pubertad |
SIA: síndrome de insensibilidad a los andrógenos.
Los grados 6 y 7 de ambigüedad sexual, propuestos por Quigley9 no se distinguen en la etapa prepuberal.
Paciente mujer de 53 años de edad que refiere desarrollo puberal con ciclos menstruales irregulares, niega abortos, cursa con infertilidad. Tiene antecedentes quirúrgicos de escisión de tumor inguinal izquierdo. Es referida a nuestra institución con un reporte histopatológico de tumor germinal de ovario izquierdo del tipo disgerminoma.
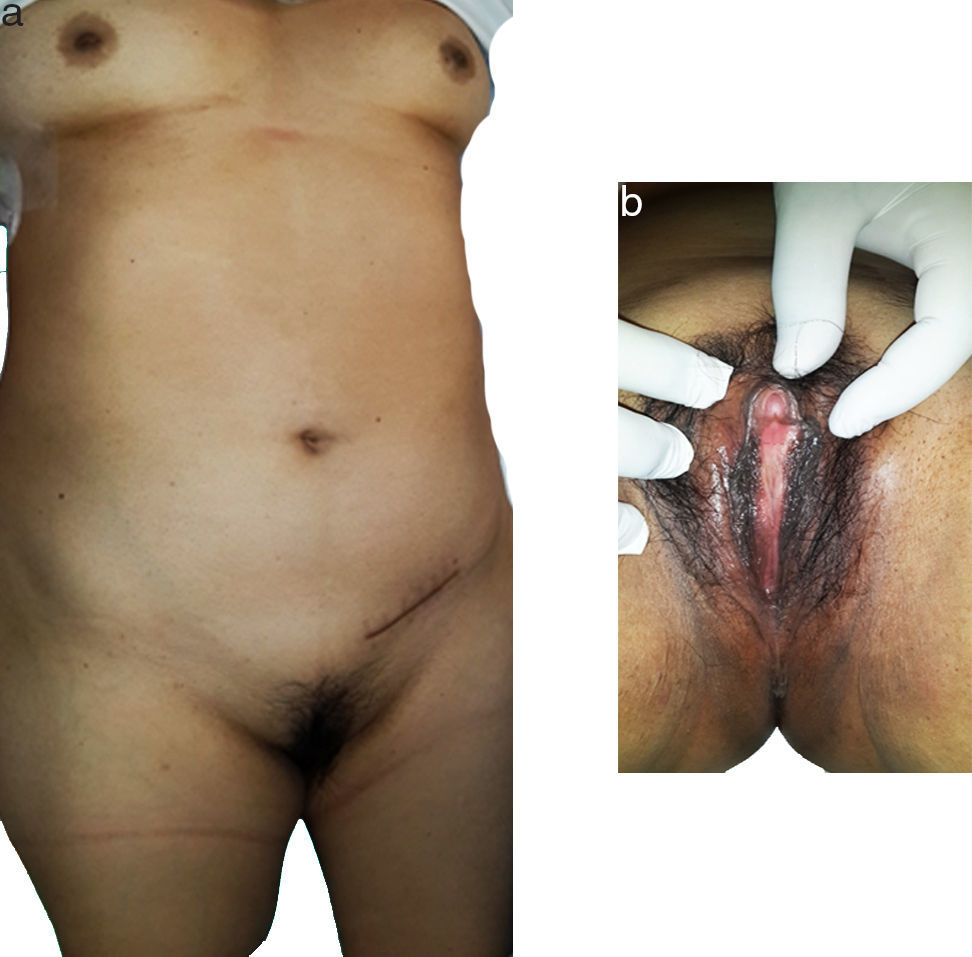
Al examen físico no muestra facies patológica, tiene presencia de glándulas mamarias de tipo femenino y genitales externos femeninos normales, pelo axilar escaso así como una lesión tumoral palpable en ingle derecha (fig. 1). Al tacto vaginal tiene ausencia de cérvix y no se palpa útero.
 Se observa el desarrollo mamario, disposición del vello ginecoide así como la herida de la cirugía previa en región inguinal izquierda. b) Se observan los genitales externos femeninos.")
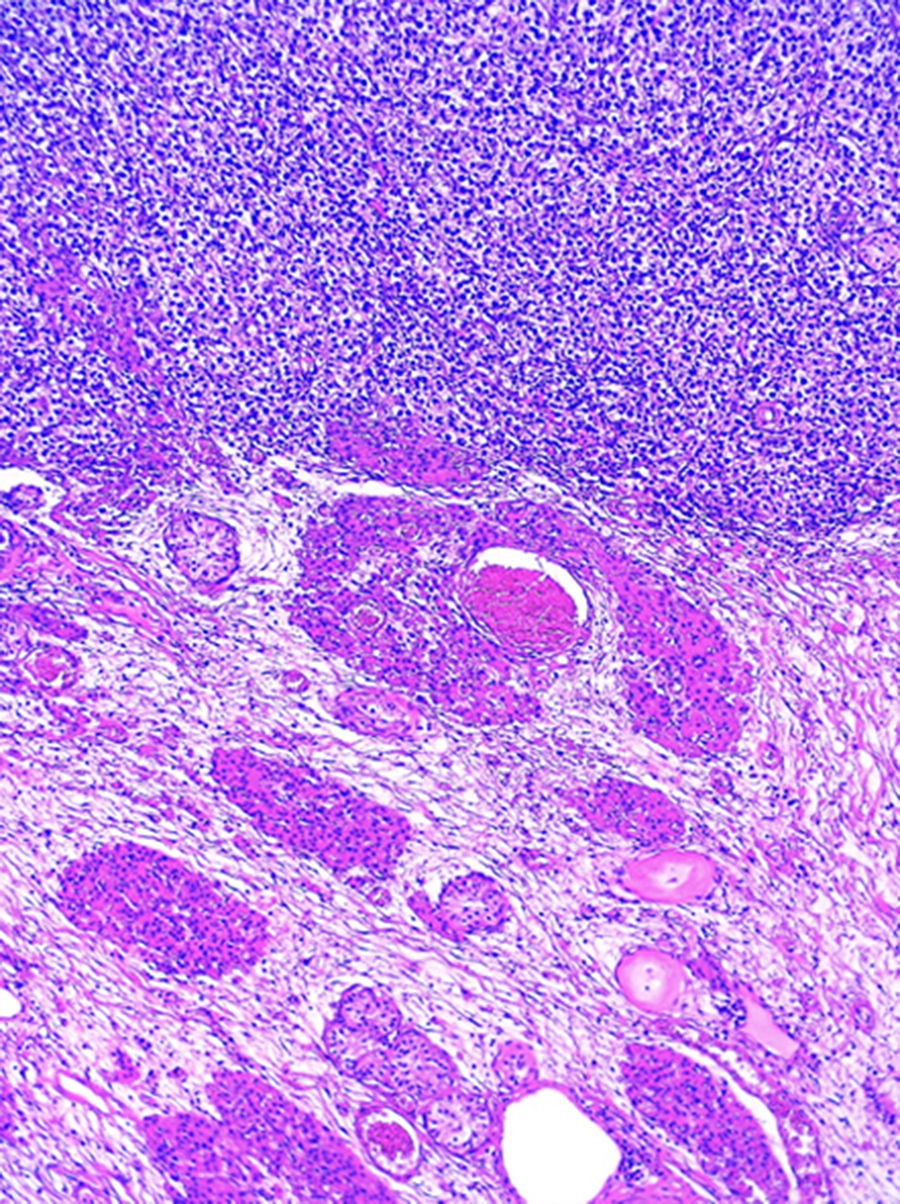
Al estudio histopatológico de los bloques y laminillas referidos como «ovario izquierdo» se observó parénquima testicular con atrofia y una neoplasia germinal tipo seminoma clásico puro (fig. 2). En el testículo residual se identificó neoplasia intratubular germinal focal.
, con la presencia de un seminoma clásico puro (parte superior de la fotografía). (Hematoxilina y eosina, X100).")
Los cortes histológicos de la resección de la lesión inguinal izquierda demuestran: parénquima testicular con atrofia, hiperplasia de células de Leydig y edema (parte inferior), con la presencia de un seminoma clásico puro (parte superior de la fotografía). (Hematoxilina y eosina, X100).
Con relación al estado hormonal de la paciente, este se presentó similar a lo descrito por otros autores, con un nivel de testosterona en suero antes de intervención quirúrgica de 389.00ng/dL (masculino normal, 240-950ng/dL), de la hormona luteinizante 31,531.50mU/mL (masculino normal, del 1 al 10mU/mL). Gonadotropina coriónica humana de 10.60U/mL (normal 5U/mL) y la alfafetoproteína de 2.77ng/mL (normal 15ng/mL) antígeno carcinoembrionario de 1.19ng/mL, CA-125 de 7.49U/ml y DHL de 213UI/l (rango normal 114 a198UI/L).
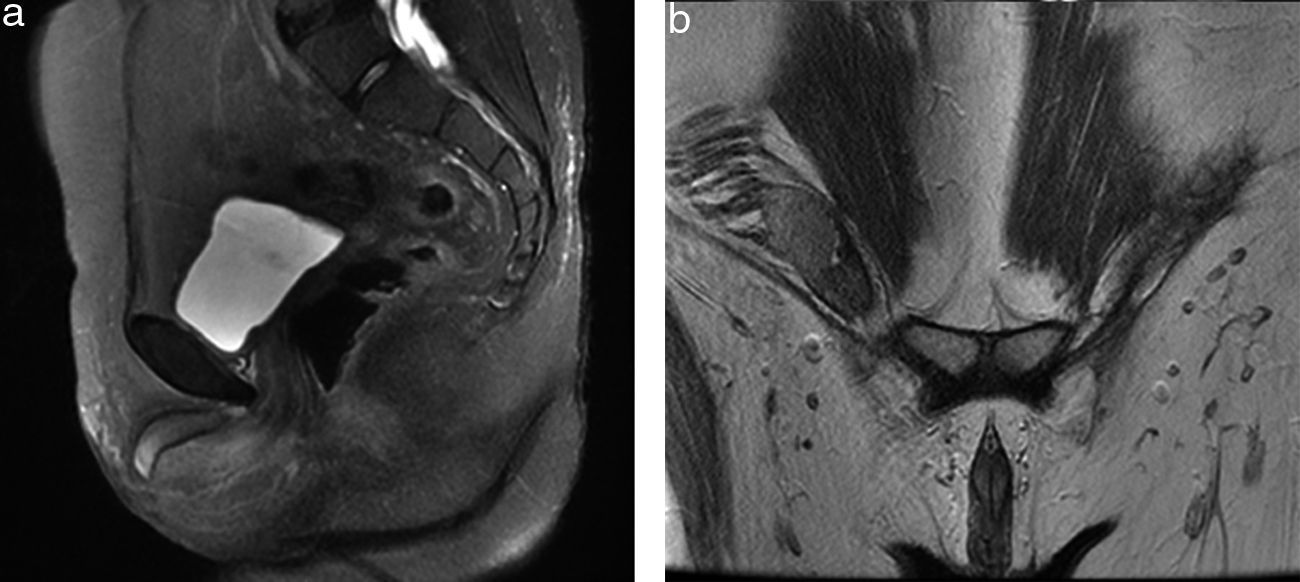
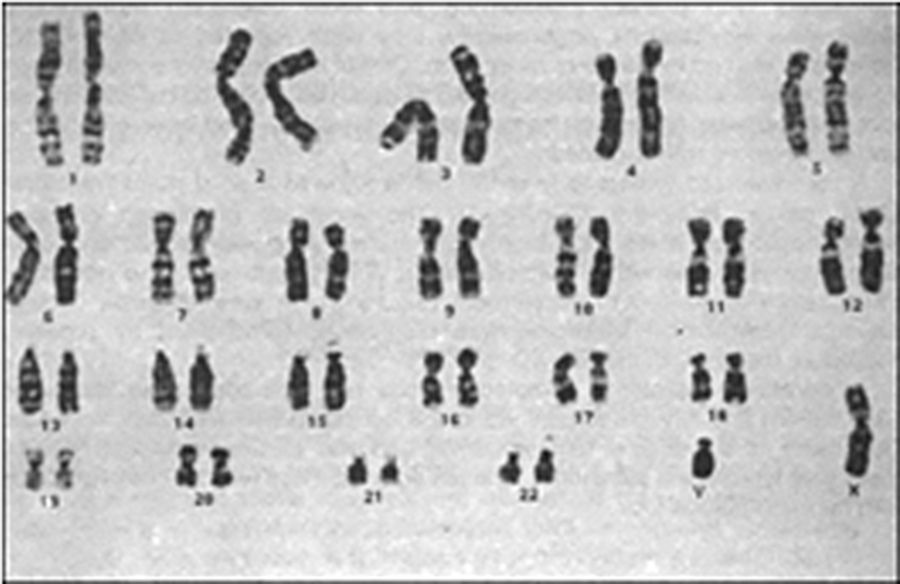
Se le realizó una tomografía computada en fase contrastada, en plano coronal y plano axial, donde se documentó un conglomerado ganglionar retroperitoneal de localización paraaórtica, el cual se extiende inmediatamente por debajo de la vena renal, criptorquidia derecha a través del anillo inguinal interno y cambios posquirúrgicos en la región inguinal izquierda (fig. 3). El análisis cromosómico y el cariotipo demostró 46XY (fig. 4).
 en plano sagital; b) en plano coronal, donde se observa la ausencia del útero y ambos ovarios, y se evidencia la presencia del testículo derecho dentro del canal inguinal ipsilateral, así como presencia de cuerpos cavernosos y vestigio del tubérculo genital.")
Resonancia magnética de pelvis. Secuencia potenciada en T2 a) en plano sagital; b) en plano coronal, donde se observa la ausencia del útero y ambos ovarios, y se evidencia la presencia del testículo derecho dentro del canal inguinal ipsilateral, así como presencia de cuerpos cavernosos y vestigio del tubérculo genital.

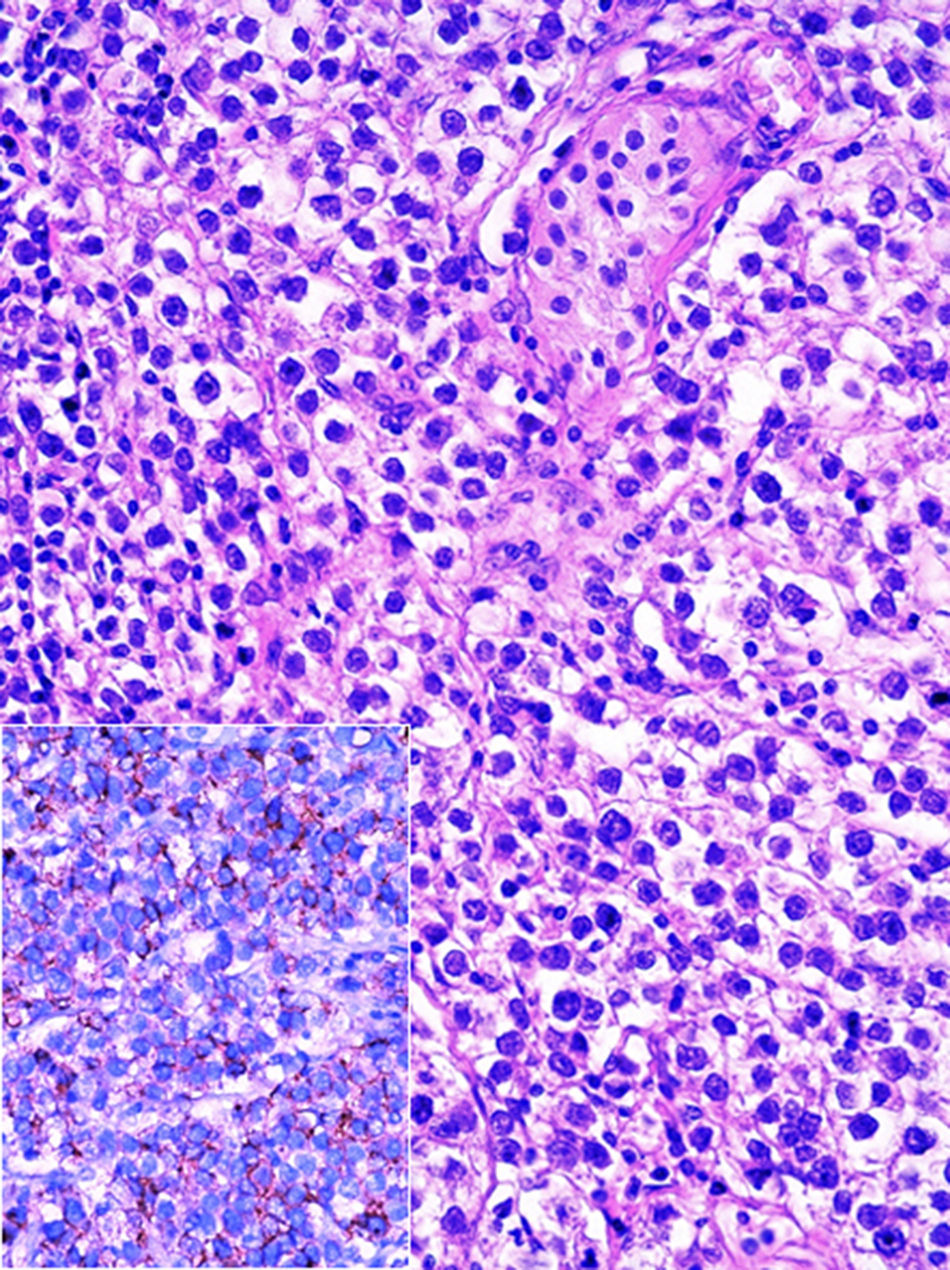
En sesión interdisciplinaria se decidió tratar mediante cirugía. El producto de la orquiectomía derecha midió 4.6×2.2×2cm, constituido por testículo, epidídimo y cordón espermático. Al estudio histopatológico se observó parénquima testicular con atrofia, hiperplasia de células de Leydig y microcalcificaciones (testículo criptorquídico), así como focos de neoplasia intratubular germinal, los cuales resultaron positivos para PLAP (fig. 5) y C-kit. La túnica vaginalis y el cordón espermático se encontraron libres de neoplasia.
 y en inserto PLAP (fosfatasa alcalina placentaria, reacción de inmunohistoquímica, X400).")
La evolución postoperatoria de la paciente y su seguimiento han sido favorables, sin datos de recurrencia de la enfermedad a 12 meses de la cirugía.
DiscusiónPresentamos el caso de una paciente con cariotipo 46XY, que corresponde a un caso confirmado de síndrome de insensibilidad a los andrógenos, quien desarrolló neoplasias en ambos testículos no descendidos, un seminoma clásico puro en un testículo y en el contralateral, la lesión precursora: una neoplasia intratubular germinal.
El síndrome de insensibilidad a los andrógenos se trata de una condición que se presenta clásicamente en la adolescencia como: amenorrea primaria o como una masa inguinal en la infancia, que es a menudo el resultado de un testículo no descendido10,11. El tratamiento es con orquiectomía bilateral y terapia de reemplazo hormonal posterior, ya que a mayor edad se asocia un mayor riesgo de malignidad testicular12. El apoyo psicológico y asesoramiento también son esenciales, tanto para los pacientes como para sus familias. Para la evaluación del grado de ambigüedad sexual se aplican los criterios descritos por Quigley9.
Más de 800 mutaciones en el gen antígeno receptor se han reportado en pacientes con síndrome de insensibilidad a los andrógenos, que se distribuyen por todo el gen con una preponderancia situada en el dominio de unión al ligando. Las mutaciones más severas están generalmente asociadas con un fenotipo síndrome de insensibilidad a los andrógenos completo, pero la correlación está menos definida en el síndrome de insensibilidad a los andrógenos parcial.
El síndrome de insensibilidad a los andrógenos constituye una entidad con un espectro clínico que va desde el fenotipo femenino, como es el caso que presentamos, hasta formas leves de ambigüedad sexual que amerita estudio genético, hormonal y de diagnóstico por imágenes, los cuales deberían beneficiar a los pacientes al igual que a sus familiares, dado que esta entidad es un trastorno hereditario de la receptividad a los andrógenos. Su tratamiento debe ser abordado por un equipo multidisciplinario, con evaluación de funcionalidad genital y de la identidad sexual para su tratamiento, en el cual la opinión de la familia y del paciente son cruciales. A través de este caso, hacemos hincapié en la asociación del síndrome de insensibilidad a los andrógenos y una tumoración inguinal que pone de relieve el riesgo de malignidad testicular. Los 2 tipos más frecuentes de tumores testiculares relacionados con síndrome de insensibilidad a los andrógenos son los de células germinales y de células de Sertoli. El factor de riesgo principal para el desarrollo de dichas neoplasias es la ausencia de descenso testicular, que representan el 10% de los pacientes con tumores testiculares13.
La tasa de síndrome de insensibilidad a los andrógenos en las mujeres con diagnóstico de hernia es 1.6%14. Unos pocos estudios retrospectivos han estimado que del 0.8 al 2.4% de las niñas con las hernias inguinales tienen síndrome de insensibilidad a los andrógenos15. Se han publicado casos de carácter familiar en los que algunos miembros muestran defectos mínimos en la virilización (microfalo o escroto bífido) y anomalías más severas, tales como: hipospadias perineoescrotal, ausencia de vasa deferens y orificio vaginal16,17. Un estudio de 150 casos18 identificó mutaciones en el 39% de los pacientes y el establecimiento de la causa de la patogénesis en el 60% de ellos, con presentación tardía en la mayoría, y con poca concientización de los afectados y sus familiares.
El diagnóstico de síndrome de insensibilidad a los andrógenos se retrasó en esta paciente. La reflexión obliga a concientizar el examen físico cuidadoso y exploración ginecológica detallada en todas las pacientes con amenorrea primaria, infertilidad y tumoración inguinal. En el pasado, creemos, muchos casos de masas inguinales fueron considerados como tejido ectópico, fibromas, leiomiomas o neoplasias benignas de origen mesenquimal, pero es claro que los pacientes deben saber si el procedimiento fue una reparación de la hernia directa o si se ha encontrado una gónada y por tanto, si se ha eliminado. En nuestro caso, parece que la historia de la cirugía inguinal izquierda condujo a la errónea suposición de que se realizó una plastia de hernia inguinal. Esto demuestra las graves consecuencias de hacer suposiciones con respecto a la gestión de los pacientes con historial médico pasado vago, que no demuestran claramente la realización de orquiectomía. La revisión histopatológica confirmó que la neoplasia de tejido extraído del conducto inguinal corresponde a un testículo izquierdo y se observó el derecho por tomografía axial computada, se extirpó posteriormente. Se encontró la lesión precursora de un tumor germinal, la neoplasia intratubular germinal, que si no se hubiera diagnosticado y extirpado, probablemente hubiera desarrollado otro tumor.
La tasa de supervivencia a 5 años del tumor testicular de bajo grado corresponde al 90-95%13, por lo que la expectativa de vida y el pronóstico de esta paciente es bueno.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.