


El hepatocolangiocarcinoma es una entidad poco frecuente de tumor primitivo hepático que muestra diferenciación tanto hepatocelular como del epitelio biliar. Su diagnóstico suele ser tardío, ya que se presenta en pacientes jóvenes, sin enfermedades asociadas y con síntomas inespecíficos. La mayoría de los casos se confunde con otro tipo de carcinoma, sobre todo con el hepatocarcinoma fibrolamelar, por ser más frecuente y presentar características clínicas y radiológicas similares.
Caso clínicoPresentamos el caso de una paciente de 26 años con un hepatocolangiocarcinoma gigante, con dificultades en su diagnóstico y un complicado abordaje quirúrgico.
DiscusiónEl diagnóstico definitivo de esta enfermedad se define por la demostración histológica de una diferenciación dual hepatocelular y colangiolar. El tratamiento quirúrgico es siempre de elección, con una supervivencia inferior a la del hepatocarcinoma y de los colangiocarcinomas clásicos. En algunos pacientes con factores pronósticos desfavorables se puede dar adyuvancia con quimioterapia, dirigida principalmente al componente colangiolar.
ConclusiónLa incidencia real del hepatocolangiocarcinoma varía entre el 2 y el 5% de los casos, y es uno de los tipos histológicos más raros que existen. El gran tamaño y la hipervascularización de la tumoración dificultan el abordaje quirúrgico en este tipo de pacientes, mientras que las características histológicas, poco frecuentes, requieren un estudio más detallado de la pieza y la aplicación de técnicas inmunohistoquímicas que confirmen el diagnóstico.
Combined hepatocellular-cholangiocarcinoma is a rare primary hepatic tumour, showing both hepatocellular as well as biliary epithelium differentiation. Its diagnosis is often delayed, as it occurs in young patients without comorbidities and with non-specific symptoms. Most cases are confused with other types of cancer, especially fibrolamellar liver cancer, which is more frequent and has similar clinical and radiological features.
Clinical caseThe case is presented of a 26 year old woman with a giant combined hepatocellular-cholangiocarcinoma with difficulties in its diagnosis and a complicated surgical approach.
DiscussionThe definitive diagnosis of this disease is defined by the histological demonstration of cholangiolar and hepatocellular differentiation, with surgical treatment always being the best choice, but with lower survival than classic hepatocellular carcinoma and cholangiocarcinoma. In some patients with unfavourable prognostic factors, adjuvant chemotherapy mainly directed cholangiolar component can be given.
ConclusionThe current incidence of combined hepatocellular-cholangiocarcinoma varies from 2 to 5% of cases, and is one of the rarest histological types in the world. The large size and hypervascularisation of the tumour makes a surgical approach difficult in these patients, while the rare histological features require a more detailed study of the piece and the application of immunohistochemical techniques to confirm the diagnosis.
El hepatocolangiocarcinoma es una entidad poco frecuente de tumor primitivo hepático que muestra diferenciación tanto hepatocelular como del epitelio biliar1. Su diagnóstico suele ser tardío, ya que se presenta en pacientes jóvenes, sin enfermedades asociadas y con síntomas inespecíficos1,2. La mayoría de los casos se confunde con otro tipo de carcinoma3, sobre todo con el hepatocarcinoma fibrolamelar, por ser más frecuente y presentar características clínicas y radiológicas similares4.
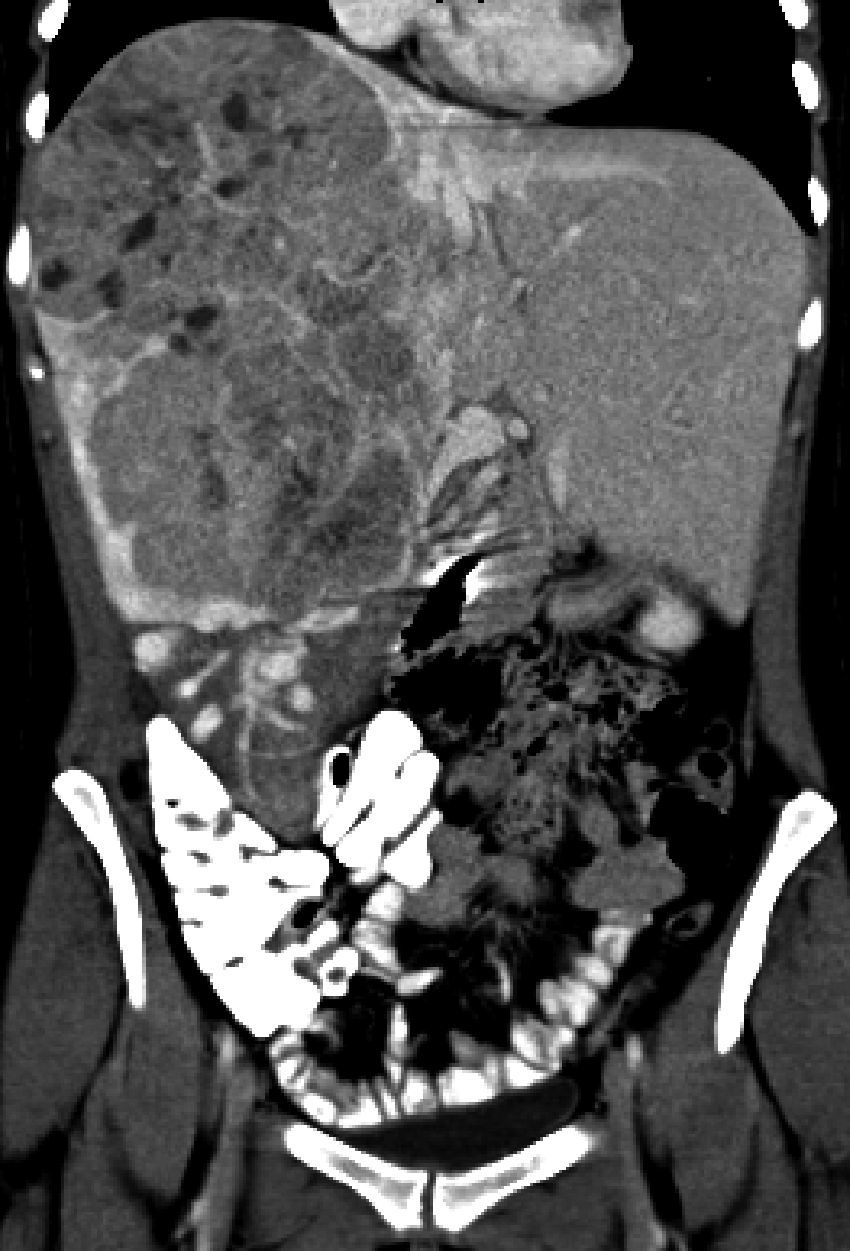
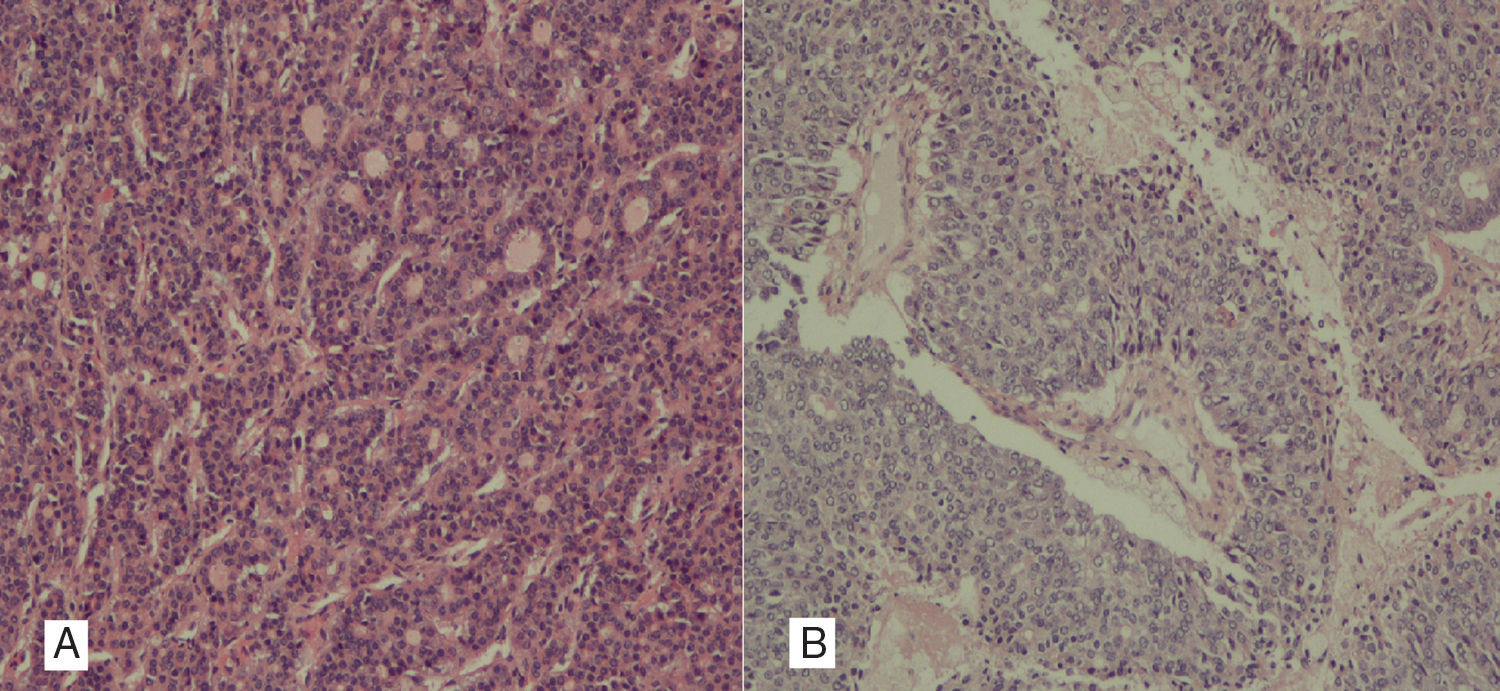
Caso clínicoPaciente mujer de 26 años sin antecedentes de interés, que presenta dolor abdominal y diarreas de un año de evolución con masa palpable a la exploración física. Se realiza tomografía axial computada abdominopélvica, y se observa masa hepática de 20×20×14cm que ocupa el lóbulo derecho (segmentos viii, vii) y parcialmente el vi, de contornos lobulados bien delimitados, con realce arterial y lavado en la fase portal, con áreas focales hipodensas por necrosis. También se observan hallazgos radiológicos sugestivos de hepatocarcinoma fibrolamelar (fig. 1). La lesión se encuentra en íntimo contacto con la vena suprahepática izquierda, probablemente invadiendo el segmento IVa. Existe muy marcada circulación colateral de tipo venoso que discurre por los segmentos vi y v, que drena en las venas hepáticas izquierda y derecha. Se realiza abordaje quirúrgico y se obtiene una tumoración gigante hepática, que asienta y sustituye todo el lóbulo derecho hepático de 25×18cm, sobrepasa la línea del ligamento redondo, llega hasta el segmento ii y ocupa prácticamente todo el segmento iv, con hipervascularización en todos los territorios. La ecografía intraoperatoria descarta otras lesiones satélites. Se realiza abordaje anterior y transección hepática con dificultad, por amplia vascularización neoformada que produce un sangrado importante, que requiere 6 pinzamientos, con un tiempo total de 1h y 47m. Se completa la triseccionectomía derecha, con pequeña ampliación al segmento ii. La paciente evoluciona favorablemente con anemización moderada que no requiere transfusiones y en el control radiológico no se evidencian complicaciones; persiste líquido ascítico en lecho quirúrgico que es controlado con diuréticos; fue dada de alta a la semana de la intervención. Actualmente, 7 meses después de la cirugía, se encuentra asintomática y sin datos de actividad tumoral. En el estudio histológico definitivo la lesión mide 14.7×9cm con márgenes libres y se observa un carcinoma mixto hepatobiliar o hepatocolangiocarcinoma combinado, constituido por los 2 tipos tumorales, hepatocarcinoma y colangiocarcinoma, en áreas claramente distinguibles, sin un área de transición. Se trata de un hepatocolangiocarcinoma mixto, de tipo clásico de OMS con los 2 patrones morfológicos e inmunohistoquímicos. El hepatocarcinoma está constituido por células tumorales en disposición trabecular y glandular, mientras que el colangiocarcinoma es un tumor desmoplásico, tubuloglandular, más indiferenciado. Además, las técnicas de inmunohistoquímica realizadas muestran en algunas zonas de la tumoración positividad para Hepatocyte paraffin 1 (Hep-Par1) marcador correspondiente al componente de tipo hepatocarcinoma (fig. 2A) y en otras, positividad para la citoqueratina AE1-AE3, 7 y villina hallazgos típicos de los colangiocarcinomas (fig. 2B).
 y parcialmente el vi de contornos lobulados bien delimitados, con realce arterial y lavado en la fase portal. Persisten áreas focales hipodensas por necrosis. Hallazgos radiológicos sugestivos de hepatocarcinoma fibrolamelar.")
Tomografía axial computada abdominopélvica en fase arterial, masa hepática de 20×20×14cm que ocupa el lóbulo derecho (segmentos viii, vii) y parcialmente el vi de contornos lobulados bien delimitados, con realce arterial y lavado en la fase portal. Persisten áreas focales hipodensas por necrosis. Hallazgos radiológicos sugestivos de hepatocarcinoma fibrolamelar.
 Imagen histológica con patrón pseudoglandular de hepatocarcinoma, con formación de luces glandulares. Hepatocyte paraffin 1 (Hep-Par1) positivo, citoqueratinas 7 y 20 negativas. B) Colangiocarcinoma poco diferenciado, en el que se distinguen luces glandulares y zonas de necrosis intratumoral. Citoqueratinas AE1-AE3 y 7 positivas; el Hep-Par1 y la citoqueratina 20, negativos.")
A) Imagen histológica con patrón pseudoglandular de hepatocarcinoma, con formación de luces glandulares. Hepatocyte paraffin 1 (Hep-Par1) positivo, citoqueratinas 7 y 20 negativas. B) Colangiocarcinoma poco diferenciado, en el que se distinguen luces glandulares y zonas de necrosis intratumoral. Citoqueratinas AE1-AE3 y 7 positivas; el Hep-Par1 y la citoqueratina 20, negativos.
La incidencia real del hepatocolangiocarcinoma es desconocida pero varía entre el 2 y el 5% de los casos1–3; es uno de los tipos histológicos más raros que existen. Su histogénesis no está clara, pero probablemente se origine de células de tipo intermedio o células progenitoras de tipo dual, sin tener relación con la presencia de cirrosis hepática, lo que lo diferencia del hepatocarcinoma2. Existen factores de riesgo que contribuyen al desarrollo de este carcinoma, como la infección por virus de la hepatitis B, el consumo excesivo de alcohol, historia familiar de cáncer hepático y diabetes mellitus, sin tener relación con la infección por virus de la hepatitis C5.
La presentación clínica suele ser igual a las del hepatocarcinoma en la que es necesario un diagnóstico diferencial con otros tumores que asientan sobre tejido hepático sano o con enfermedades hepáticas benignas como la hiperplasia nodular focal6. El diagnóstico definitivo se define por la demostración histológica de una diferenciación dual hepatocelular y colangiolar, la cual puede ser de 3 tipos: doble, cuando existe presencia de hepatocarcinoma y colangiocarcinoma en el mismo hígado pero separados; combinada, cuando ambos carcinomas se encuentran contiguos y confluyen al crecer; o mixta, cuando se encuentran presentes en una única masa tumoral íntimamente entremezclada3,7. La clasificación reciente de la OMS unifica las anteriores en un grupo de tipo clásico y las diferencia de otro grupo dividido según la presencia de células intermedias con distintos inmunofenotipos de células madres8. El grado histológico de malignidad se divide utilizando una escala del i al iv de acuerdo con el incremento de las irregularidades nucleares, del hipercromatismo y de la relación núcleo/citoplasma4. En nuestro caso se clasificaría como un tipo clásico de OMS (áreas de hepatocarcinoma y colangiocarcinoma típicas)8 y un tipo B de Allen y Lisa (tipo combinado o de colisión, que son contiguos por que confluyen al crecer, pero sin observar áreas intermedias)7. El estudio inmunohistoquímico puede aclarar dudas diagnósticas y confirmar el componente de carcinoma colangiolar con la positividad del antígeno carcinoembrionario, la citoqueratina 7 y el CA19-9, y la del hepatocarcinoma con la positividad del Hep-Par1 y la alfafetoproteína. Ambos deben estar presentes en al menos el 10% del tumor para considerarlo hepatocolangiocarcinoma2,3.
ConclusiónEl tratamiento quirúrgico del hepatocolangiocarcinoma es siempre de elección, con una supervivencia inferior a las del hepatocarcinoma y colangiocarcinomas clásicos2,9. En algunos pacientes con factores pronósticos desfavorables se puede dar adyuvancia con quimioterapia, dirigida principalmente al componente colangiolar, que es el más relacionado con la recidiva y la presencia de enfermedad a distancia10. El gran tamaño y la hipervascularización de la tumoración dificultan el abordaje quirúrgico en este tipo de pacientes y este es el único tratamiento efectivo actualmente, mientras que las características histológicas poco frecuentes requieren un estudio más detallado de la pieza y la aplicación de técnicas inmunohistoquímicas que confirmen el diagnóstico.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.