


Thyroid cancer is the most common malignancy of the endocrine system, the papillary variant accounts for 80–90% of all diagnosed cases. In the development of papillary thyroid cancer, BRAF and RAS genes are mainly affected, resulting in a modification of the system of intracellular signalling proteins known as “protein kinase mitogen-activated” (MAPK) which consist of “modules” of internal signalling proteins (Receptor/Ras/Raf/MEK/ERK) from the cell membrane to the nucleus. In thyroid cancer, these signanling proteins regulate diverse cellular processes such as differentiation, growth, development and apoptosis. MAPK play an important role in the pathogenesis of thyroid cancer as they are used as molecular biomarkers for diagnostic, prognostic and as possible therapeutic molecular targets. Mutations in BRAF gene have been correlated with poor response to treatment with traditional chemotherapy and as an indicator of poor prognosis.
ObjectiveTo review the molecular mechanisms involved in intracellular signalling of BRAF and RAS genes in thyroid cancer.
ConclusionsMolecular therapy research is in progress for this type of cancer as new molecules have been developed in order to inhibit any of the components of the signalling pathway (RET/PTC)/Ras/Raf/MEK/ERK; with special emphasis on the (RET/PTC)/Ras/Raf section, which is a major effector of ERK pathway.
El cáncer de tiroides es el tumour maligno más frecuente del sistema endocrino; la variante papilar representa entre el 80 y el 90% de todos los casos diagnosticados. En el desarrollo del cáncer papilar de tiroides están afectados principalmente 2genes llamados BRAF y RAS, que alteran el sistema de señalización intracelular de las proteínas conocidas como «cinasas de proteínas activadas por mitógenos» (MAPK, por sus siglas en inglés para mitogen-activated protein kinase) y que se componen de «módulos» de proteínas de señalización interna (Receptor/Ras/Raf/MEK/ERK), que van de la membrana celular al núcleo y que en el cáncer de tiroides regulan diversos procesos celulares, como la diferenciación, crecimiento, desarrollo y apoptosis. Tienen un papel importante en la patogénesis del cáncer de tiroides, debido a que son usados como biomarcadores moleculares, como elementos de diagnóstico, pronóstico y como posibles blancos terapéuticos moleculares.
ObjetivoRevisar los mecanismos moleculares que intervienen en las vías de señalización, en las que están involucradas las proteínas de los genes BRAF y RAS, en el cáncer de tiroides.
ConclusionesLas mutaciones en el gen BRAF han sido correlacionadas con una pobre respuesta al tratamiento con quimioterapia tradicional, además de ser un índice de mal pronóstico. La terapia molecular es de gran interés en este tipo de cáncer, ya que se han desarrollado medicamentos que actúan inhibiendo alguno de los componentes de la vía de señalización (RET/PTC)/Ras/Raf/MEK/ERK, con especial énfasis en la sección (RET/PTC)/Ras/Raf, que constituye un efector principal de la vía ERK.
Thyroid cancer is the most common malignancy of the endocrine system, and represents approximately 1% of cancer cases diagnosed worldwide. Differentiated thyroid cancer includes papillary and follicular types. The most frequent is papillary carcinoma, which accounts for approximately 80–90% of all cases diagnosed and most frequently presents in women rather than men, at a 2:1 ratio. Its incidence worldwide has significantly increased during the last 3 decades, probably due to more advanced systems of diagnosis, which has resulted in more timely diagnoses and increasingly more targeted treatments.1–5 Recent reports indicate that there is a similar prevalence in diagnostic ages, regardless of ethnic group. However, there are differences in prevalence by gender and ethnicity. It has been reported that Caucasian men and women present a prevalence of 6.3% and 7.1%; in the English population prevalence is reported at 4.3% and 8.4%; Hispanic at 4.2% and 6.7% and Asian at 3.4% and 6.4%, respectively.6 It is believed that genetic factors, environmental influences and access to health services may be factors which determine the incidence of thyroid cancer in any one particular region. However, during the last decade a sustained increase in the rate of thyroid cancer worldwide has been observed, mainly of the papillary type.6
3195 cases of thyroid cancer were reported in Mexico in 2008 (1351 male and 1844 female), which represented 2.5% of total malignancies, with an incidence of 3 out of every 100,000 inhabitants and a mortality of 0.6 per 100,000 inhabitants, becoming the sixth causes of death in women and the thirteenth cause of death in men, and with maximum frequency of between 41 and 50 years of age: 60% of cases occur between 31 and 60. With regards to thyroid cancers, papillary carcinoma and its variants represent 80.3% and follicular cancer and its variants, 2.4%.7–9
On a molecular level, papillary thyroid cancer frequently presents with metabolic changes in the intracellular signalling systems, which involve the activation of proteins known as mitogen-activated protein kinase (MAPK). The latter have, in recent years, been the purpose of many studies due to their role in the pathogenesis of thyroid cancer and their use as molecular biomarkers, in addition to their potential usage for diagnostic, prognostic purposes and as possible therapeutic molecular targets.10–12
Genetic changes in cancerIt is a known fact that changes in 3 types of genes may be responsible for the start and continuation of the cancer: (a) oncogenes, (b) tumour suppressor genes and (c) stability genes. It is also well known that the cell has several mechanisms to safeguard and protect the organism from a lethal, potentially carcinogenic effect such as a genetic mutation and it is therefore only when there are changes in several genes that cancer may develop.12–14
A mutation in an oncogene leads to a gene which should remain constituitively active or inactive normally becoming inactive or active. The activation of an oncogene may be due to: chromosomal translocation, abnormal gene amplification or intragenic mutation which affects a vital residual expression for the activity of this gene. A mutation of an oncogene allele generally suffices to confer lack of control in cellular growth. For their part, tumour suppressor genes act against what may be a genetic mutation; in their case, they reduce the gene's activity and its product. This inactivation is a product of the mutations which lead to the loss of essential residual expressions for the activity of a protein coded by this gene, which in turn results in truncated proteins, albeit due to deletion, insertion or epigenetic alterations. The third type of cancer genes are called stability genes or “watchdog” genes. In their case, this type of genes promote tumour genesis via different routes to the other 2 types of tumour genes mentioned previously. This type of “watchdog” genes are the ones which are mainly engaged in repairing all changes resulting from DNA replication when this occurs normally or is exposed to a mutagen. Other stability genes also control the processes which involve large quantities of chromosomes, such as those which regulate chromosomal recombination and segregation which occur during mitosis. The stabilising genes maintain genetic alterations to a minimum, and when they are inactive, there is an increase in the rate of mutations.14–17
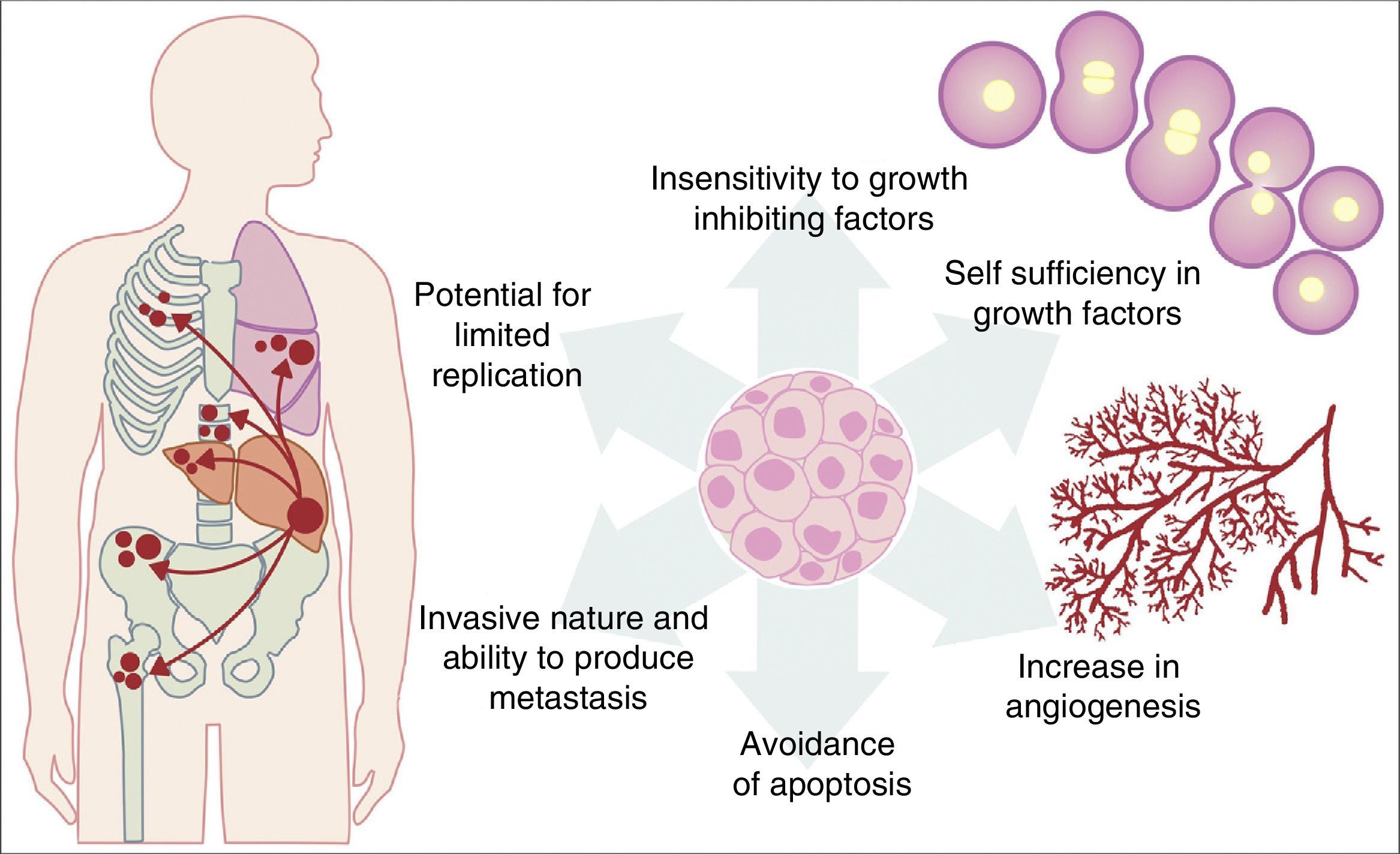
All the previously described mutations function in the same manner as a normal cellular physiological process. Cancer cell growth may be provoked by the activation of oncogenes, which control the cellular cycle on inhibiting cellular death (apoptosis) or increasing the transition of cells of the G0 to G1 stage (“release of cell arrest”) or helping to supply nutrients when phenomena such as angiogenesis increase. It is important to remember that a mutation is defined as any change in the sequence of the genome and that these changes include those which influence a pair of bases, and those which are minor or major changes such as: deletions, insertions, amplifications or translocations. All types of mutations may present in cancer cells but they are considered to be a disease when occurrence of a mutation posterior to development is pathogenic.14–17 Furthermore, once the cell has become cancerous, it presents with a series of metabolic, molecular and structural changes which allow it to survive, develop and even metastasise. Among these general changes may be found: self-sufficiency in growth factors, lack of sensitivity of inhibiting growth factors, a capacity for invasion into nearby tissues and metastasis, a potential for unlimited replication, an increase in angiogenesis and the development of mechanisms of apoptosis evasion (Fig. 1).18 The Knudson hypothesis has considerably contributed to the understanding of how mutations in genes involved in the development of cancer work, where there is a dominant autosomic heritage, due to the loss of function of a tumour suppressor gene, leading to the subsequent tissue-specific mutation. However, these contributions were prior to the increasing knowledge we now have regarding the regulation of the non coding micro-RNA and RNA which has led to the “continuous model “of tumour suppressor gene regulation where not only hereditary genetic and somatic mutations contribute to the development of cancer, but also the epigenetic elements, such as the microRNA elements which significantly contribute to the development of cancer.19

Table illustrating some of the pathological characteristics acquired by a cancer cell which has helped it to survive in the body.
In thyroid cancer there are a number of genetic alterations which are related to cellular progression and differentiation. These alterations may be summarised in 2 main categories: (1) chromosomal rearranging and (2) specific mutation. Both alterations primarily influence 2 genes called BRAF and RAS. The mutations of several of these 2 genes have been found in over 80% of cases with papillary thyroid cancer and rarely overlap in the same tumour. These mutations change the transduction system of intracellular signalling, which regulates several metabolic pathways, and this has direct repercussions on cellular processes such as differentiation, expansion and apoptosis, among others. In accordance with current data, mutations which affect BRAF exclusively exist in papillary thyroid cancer and in anaplasic thyroid cancer, but have not been reported in other histological types of thyroid cancer, such as follicular thyroid cancer and medullary thyroid cancer. Furthermore, it has been reported that the BRAFV600E mutation (replacement of a valine by a glutamic acid in position number 600) is found in almost 60% of all types of thyroid cancers. The mutations lead to activation in the MAP-kinase-dependent signalling pathways which it is believed are an early event in the development and progression of thyroid cancer.20,21
The cascade of MAP-kinase-dependent signalling pathway processes is very important, since these processes regulate the number and participation of genes involved in proliferation, differentiation and survival of cancer cells. The MAP kinase system is composed of protein signalling “modules”, which extend from the cell membrane to the nucleus and which are preserved from yeasts to vertebrates.22–24 This system is regulated by a cascade of phosphorylations where other phosphorylases intervene, located in 2 places in relation to the MAP kinase: (1) “Upstream” and (2) “downstream”.20–25
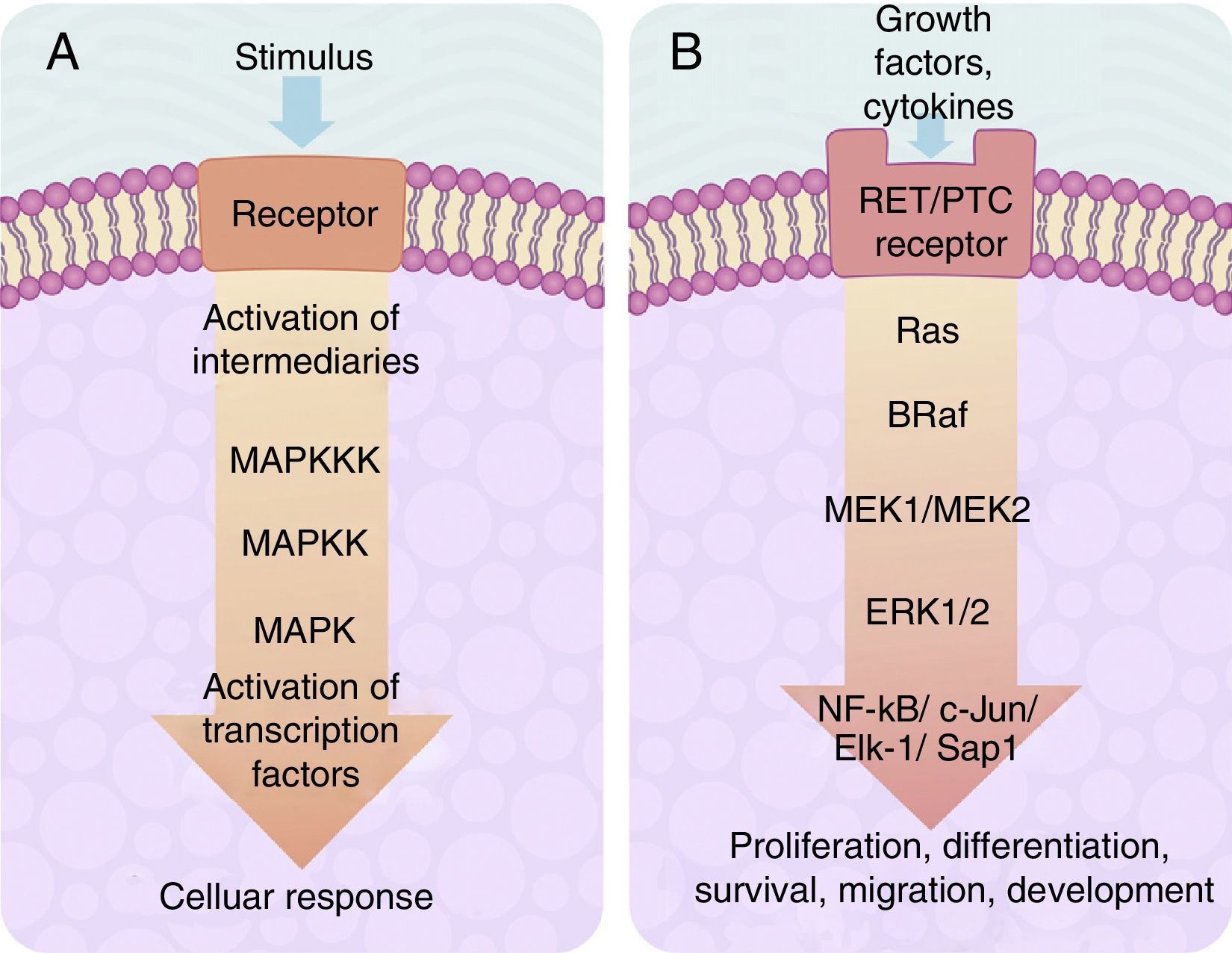
The centre of activation of any metabolic signal linked through a cascade to activate a MAP kinase pathway upstream is composed of 3 levels of kinases, which are activated in a sequence through the activation signal of a membrane receptor with tyrosine kinase activity (Fig. 2A). In eukaryotes cells there are several MAP kinase-dependent signalling pathways, which are activated by different mechanisms of stimulae. At least 4 different types of MAP kinases subfamilies have been identified to date which are: ERK-1/2, JNK-1/2/3, P38-a/b/g/d and ERK-5. The ERK-1/2 MAP kinase signalling pathway is of significance in medicine and in particular for a better understanding of the molecular generation and regulation mechanisms of thyroid cancer. It is primarily activated as a response to a mitogentic stimulus, such as the activation of a receptor for a growth factor (Fig. 2B). The ERK-1/2 MAP kinase signalling pathway is initially activated upstream with the activation of a membrane receptor to which a ligand has bound. The receptor now activates a G type protein called Ras (with kinase activity), which “recruits” a MAPKKK family called Raf through the use of subsequent phosphorylations which, in turn, activates a series of MAPKK called MEK-1/2. Finally, MEK-1/2 activates ERK-1/2which is translocated inside the nucleus, where it phosphorylates transcription factors of several genes, producing the regulation of its expression. This nuclear translocation is required in response to stimulae such as growth factors, which regulate the entry into the cellular cycle or the cellular differentiation. This is why it is important in the study of cancer, which involves the before-mentioned uncontrolled cellular processes.14,26–28
 Table showing an intracellular signalling cascade activated by a membrane receptor which activates a series of kinases downstream grouped one after the other in a MAPKKK-MAPKK-MAPK sequence and terminates in the activation of a transcription factor which originates a cellular response. (B) Outline of the activation of ERK-1/2 transcription factor through the intracellular signalling of kinases in thyroid cancer. Initial activation through a tyrosincinase (RET/PTC) type membrane receptor and which continues downstream through the sequence of the Ras/B-Raf/MEK-1/2 kinases.")
(A) Table showing an intracellular signalling cascade activated by a membrane receptor which activates a series of kinases downstream grouped one after the other in a MAPKKK-MAPKK-MAPK sequence and terminates in the activation of a transcription factor which originates a cellular response. (B) Outline of the activation of ERK-1/2 transcription factor through the intracellular signalling of kinases in thyroid cancer. Initial activation through a tyrosincinase (RET/PTC) type membrane receptor and which continues downstream through the sequence of the Ras/B-Raf/MEK-1/2 kinases.
It has been considered that the tumorigenesis process requires the presence of a deregulation of the intracellular signalling processes, which involve different levels of MAP kinases cascades, where the cancer cells acquire different capacities such as: (1) ignoring signals to proliferate; (2) avoiding apoptosis; (3) becoming insensitive to antiproliferation signals; (4) acquiring an unlimited potential to replicate; (5) invading and producing metastasis to other tissues and (6) producing angioigenesis, which provides them with nutrients and vital support.18,29
It has been reported that different malignancies show up as an alteration in the intracellular signalling pathway, comprising of the cascade of activation in sequence of the Ras/Raf/MEK/ERK kinases, in which the last cascade effector (which is the ERK-kinase protein) is activated upstream by genetic mutations which may affect the different kinasees located in some of the levels of the chain reactions, starting with an over-expression of the tyrosine kinase type receptor and most frequently with mutations in the Ras and Raf proteins. The intracellular signalling cascade in a cancer cell begins with the activation of a membrane receptor (with tyrosine kinase activity) when the latter binds with its ligand or through an external stimulus (e.g. change in osmosis, oxidative stress, lack of nutrients, etc.). Once the receptor has been activated, it recruits the first effector, by means of specific phyosphylations, of a signalling cascade which is a MAPKKK which may be A-Raf, B-Raf or Raf-1. These kinases phosphylate to the following MAPKK which are MEK-1/2 and finally phosphylate to a MAPK, which is ERK-1 or ERK-2, which are in turn translocated to the nucleus to initiate activation/inhibition of specific genes. If this signalling cascade is found constitutively in all normal cells of the body, in the cancer cell each component of the chain may have mutations which provoke excessive downstream activation of their kinase, and this increases cellular activity exponentially through the activation/inhibition of specific genes. It has therefore been reported, for example, that the mutations in the Ras and Raf kinases (which are the components of the intracellular pathway composed by the Ras/Raf/MEK/ERK receptor), are found in all types of thyroid cancer, primarily in papillary thyroid cancer. This indicates that a single alteration in these molecules may be enough to lead to a malignant transformation of the thyroid cells.
It is thus known that the presence of the mutation called BRAFV600E (consistent in the presence of a valine residual, instead of a glutamic acid residual, in position 600 of the B-Raf protein kinase), the prevalence of which has been reported between 27% and 80% in different patient groups with thyroid cancer, leads to an increase of over 400 times in the activity of B-Raf, which produces an increase in the downstream activity of the ERk effectors, which increase their phosphorylant capacity and therefore the activity of diverse key genes30 for cellular metabolism, proliferation and apoptosis.
The most frequently reported mutations in papillary thyroid cancer are: re-arrangements in the RET/PTC gene (membrane receptor with tyrosine kinase activity) and B-Raf and Ras mutations (proteins with kinase activity). They are all involved in the intracellular signalling pathway which activates the nuclear ERK effector. These alterations are exclusively found in patients with papillary thyroid cancer, which proves that every separate alteration may be sufficient to lead to a malignant transformation of thyroid cells. The proto-oncogene called RET (rearranged during transfection), encodes for a membrane receptor with tyrosine kinase activity, which is expressed in a particular manner in type C parafollicuar cells in thyroid cancer, which results in aberrant proteins with different chimeric forms from the receptor. However, their expression is very low in the follicular cells. The name RET/PTC (rearranged during transfection/papillar tyroid cancer associate) has been given to these chimeric forms. To date, more than 11 different types of RET/PTC proteins have been described, where RET/PTC1 and RET/PTC3 are the most frequently found in thyroid cancer.31,32
In the cancer cell, RET/PTC leads to constituitive downstream activation of a kinase called Ras. It is now known that Ras activates a large number of molecules which act as downstream intracellular signalling systems and induce cancer cell invasion properties. It has been reported that Ras also activates reactive proteins to stress, such as Raf kinase, which is the key downstream cytosolic Ras effector.
Several isoforms of Raf have been reported: A-Raf, B-Raf and Raf-1 (also called C-Raf). Raf activation includes a series of highly regulated metabolic steps, which start with their recruitment of the internal cellular membrane by the Ras protein. Once Raf has been activated it may bind downstream with other kinases, among which are MEK-1 and MEK-2. For activation, as well as Ras being activated, the Raf isoforms require that an enzyme with protein phosphatise activity known as Src-kinase be activated. This protein phosphorylates or dephosphorylates certain amino acid residues, located within the Raf areas, which regulate its inactive/active state. Of the 3 known isoforms for Raf, isoform B-Raf is the one which presents with the highest basal level activity compared with A-Raf or Raf-1. This is very significant, since in thyroid cancer a large number of mutations in protein B-Raf have been reported, compared to that of A-Raf or Raf-1. This has led to the use of B-Raf as a tumour marker for several types of cancer, and specifically thyroid cancer. Moreover, B-Raf is the most powerful downstream activator of MEK-1 kinase, which is the ERK effector.
The MEK-1 protein is a protein kinase with dual activity, since it is able to phosphorylate serine and tyrosine residues. Once MEK-1 has been activated by B-Raf, the downstream activation of the ERK effector occurs, which is a serine/threonine kinase which phosphorylates diverse proteins, both cytosolic and nuclear. In this way, the hyperactivation of the signalling cascade which regulates ERK may lead to the arrest of the cellular cycle. However, if there is an aberration in any of the sequences of the signalling cascade, a tumour transformation of the cell may be induced. The response kinetics and their extension to diverse ligands or extracellular stimulae may induce the ERK to regulate specific diverse biological programmes such as cellular differentiation and proliferation or apoptosis.32
Intracellular PI3K/Akt/mTOR signalling in thyroid cancerIn addition to the above mentioned intracellular signalling, there is also the phosphatidylinositol -3 kinase/protein B (Atk) pathway/mammalian target, for rapamicin (mTOR), also expressed as PI3K/Akt/mTOR. This signalling pathway is involved in process such as: cellular differentiation and growth, progression of cellular cycle, endocitosis, motility, apoptosis and intermediate metabolism (related primarily to glucose uptake).33–35 In the last few years, alterations in the said pathway have been reported in several types of cancer such as thyroid, gastric, neuro-endocrine and ovarian. Furthermore, it has been observed that the activation of this pathway in cancer cells may increase resistance to cisplatin, cabo-platin and paclitaxel treatment, and as a result their study may help to understand the mechanisms of genesis and progression of the cancer, in addition to the mechanisms of tumour resistance for being a possible target, for molecular therapy and the management of thyroid cancer.33–37
Phosphatidylinositol-3 kinase (PI3K) phosphorylates groups 3-OH of the inositol ring of phosphatidylinositol groups. The sub-products of the reaction of PI3K measure the reversible nature of the cytoplasm membrane location of proteins, which contain complex lipid binding commands and an above standard increase in their activity associated with an oncogenic cellular transformation.36,37
In terms of intracellular signalling the class I PI3K may be a downstream effector of either a membrane receptor with tyrosincinase activity or a G protein attached to a receptor.37,38 However, the protein known as Akt has an approximate molecular weight of 57kDa, which is a serine/threonine kinase that belongs to the kinase family where they following may be found: protein kinase A, protein kinase G and protein kinase C. The Akt is also known as protein kinase B (PKB).37–41
The protein called mTOR is a kinase serine/threonine coded by the FRAP 1 gene and is the primary downstream effector of the complex PI3K/Akt. There are 2 different isoforms of mTOR, called mTORC1 and mTORC2. Each one is a protein multicomplex formed by several components, where the centre is the protein mTOR itself, to which several sub-units have been added. The complex mTORC1 is formed by the protein mTOR associated with the protein RAPTOR, which works as a link and positively regulates mTOR. It is also associated with 2 negative regulators which are PRAS40 and the protein DEPTOR, whilst the sub-unit mLST8, combined with the complex mTORC1, regulates its activity.39–42 The intracellular communication pathway measured by mTORC1 is involved in cellular growth and differentiation whilst the complex mTORC2 confers insensitivity to rapamicine.41,42 De-regulation of mTOR activity is correlated with the presence of hamartomas, tuberous sclerosis, Peutz–Jeghers syndrome and with several types of cancer in which the genetic changes to the regulating genes of mTOR, such as TSC1-TSC2 and LKB1, present diverse types of mutations.42–44
During bone formation, the RUNX2 gene is key for the differentiation of the osteoblastoma, and for the proliferation of chondrocytes and endochondral differentiation and hypertrophy. Its participation as the promoter of tumour in breast and prostate cancer has been reported, as well as its association with genes which control tumour invasion and metastasis, since it promotes the expression of the metalloproteinases MMP2, MMP13, MMP14. In this way, RUNX2 has been defined as a promigratory transcription, pro-invasive, and pro-angiogenic factor, in addition to participating in the initial events of tumorogenesis and leading the spread of bone metastasis.43–45
The expression and activity of RUNX2 is induced directly and indirectly by Akt. In vitro experiments made with cancer cells have shown that the activation of the PI3K/Akt intracellular communication pathway leads directly to an increase in affinity to DNA by the RUNX2 and RUNX2-dependent factors of transcription and that the mutations affecting Akt lead to a decrease in the affinity of RUNX2, which affects the progression of the cellular cycle. Indirectly, activation of the PI3K/Akt pathway regulates RUNX2 activity, since it increases its protein stability either by acting through a nuclear transcription factor called FoxO, an activator or a specific gene transcriptional repressor and the cytoplasm.46,47
Mutations and thyroid cancerA mutation of any protein involved in the intracellular signalling cascade measured by a tyrosine type receptor may determine that a normal cell becomes a cancer cell. In this context, it has been reported that around 70% of patients who present with papillary thyroid cancer have at least one mutation which affects the ERK activation cascade; the most frequent are those affecting Ras and B-Raf and, to a lesser degree, RET/PTC.
The prevalence of RET/PTC mutations in thyroid cancer is varied, since it depends on the geographic region, but it may be stated that approximately 20% of cases of thyroid cancer present these alterations, and also that it is more frequent in young people or those with a background of having undergone radiation treatment. Furthermore, the mutations in a member of the gene family which code for the protein Ras have been primarily described in follicular adenoma and thyroid carcinoma, but only described in around 10% of papillary thyroid cancer cases.33
In the case of B-Raf (which may also be expressed as BRaf) it has been observed that it is expressed in a normal manner in: blood forming cells, neurons, testicular cells and in thyroid follicular cells. Contrary to that which occurs with the mutations in A-Raf and Raf-1 which are extremely rare, mutations in B-Raf are the most frequent in papillary thyroid cancer, as well as being the second somatic mutation found in all types of cancer which affect the human being. Thus, for example, over 45% of all types of cancer which affect the human being present a mutation in B-Raf. Close to 90% of these mutations consist of a replacement of a timine for an adenine in the exon 15 of the neucleotide 1799 (c.1799T>A), which results in a mutation of B-Raf, where a valine residual is replaced by a glutamic acid residual in the protein position 600. This mutation is expressed as BRAFV600E (also expressed as B-Raf-V600E or BRAF-V600E). It has been reported that the prevalence of this mutation in papillary thyroid cancer may vary from 29% to 83%, depending on the sample studied, and has not been described in follicular thyroid cancer.48 Research studies carried out in vitro have shown that the replacement of the valine for a glutamic acid in 600 position adjacent to a threonine residual in position 599 within protein B-Raf induces similar behaviour to a phosphorylation, which makes the tertiary structure of the protein affected where the hydrophobic interactions break between segment P (the activation site of the protein by phosphorylation) and the kinase activity segment, resulting in the BRAFV600E kinase activity being 460 times higher than the native B-Raf. This raised constituitive activity of BRAFV600E produces a downstream activation of all effectors of the signalling cascade effectors up to ERK,which results in the transformation of normal cells into cancer cells, and their unnecessary proliferation of Ras protein, for activation of the signalling pathway.
It has been proven that the presence of BRAFV600E is linked to genes involved in the metabolism of iodine, especially those involved in the uptake of iodine by cells for thyroid hormone synthesis. The presence of BRAFV600E induces the thyroid cells to present changes in several of the genes involved in this process, which have been associated with a greater aggressive tumour pattern. The presence of mutations in the BRAF gene has therefore been correlated with a poor response to standard chemotherapy treatment and also is an indication of poor prognosis, since the mutation in BRAF was found both in anaplasic cells and in poorly differentiated carcinomas, which means that the molecular changes in B-Raf are produced early on in the tumorogenesis phenomenon.48
Re-arrangements in PAX8-PPARγ in thyroid cancerThe PAX8 gene is a specific thyroid transcription factor which regulates development and differentiation. Furthermore, the PPARγ gene is involved in the control of the cellular cycle, apoptosis and carcinogenesis,49 as well in adipogenesis and sensitivity to insulin.
Together PAX8 and PPARγ have a key role in thyroid cancer. The translocation between these 2 genes produces a new gene, the overexpression of the resulting PAX8/PPARγ or PPFP fusion protein alters the PPARγ function of the downstream inhibitor of the cellular proliferation and apoptosis inductor. The chromosomal translocation PAX8-PPARγ was detected in 35% of follicular thyroid cancer and anaplasic thyroid cancer.49 Furthermore, in follicular thyroid cancer the overexpression of PAX8/PPARγ is associated with the activation of a MAP kinases depending on TGFß receptors, which is closely linked to the tumorogeneis process.49,50
Molecular therapy in thyroid cancerThyroid cancer, whether it be papillary or follicular, which is well differentiated, has a fairly promising clinical behaviour pattern since it may be treated with surgery, followed by radiotherapy. However, the tumours which are undifferentiated or those which lose their capacity to capture radioactive iodine cannot be surgically removed, and are therefore tumours of poor prognosis. This type of tumours may be treated with molecular therapy, where the intracellular signalling chain (RET/PTC)/Ras/Raf/MEK/ERK is a frequent target for molecular therapy studies.
Molecular therapy is of great interest in this type of cancer as drugs have been developed which inhibit several of the components of the (RET/PTC)/Ras/Raf/MEK/ERK signalling pathway. Particular emphasis has been place on the (RET/PTC)/Ras/Raf section, as this segment of the signalling cascade is the main ERK effector. It has been reported that the ZD6474 compound, which is a RET/PTC kinase activity, has been effective in in vitro and preclinical trials to induce arrest of the cellular cycle in human papillary carcinoma, and this impedes its growth when injected in mice.51 The compounds pyrazolopyrimidine (PP1 y PP2) and sunitinib (SU12248) have also been tested in in vitro, in vivo and preclinical trials. These nullify the RET/PTC signal for eliminating the tumorigenic effect in test animals. Promising results have also been reported in phase II clinical trials ii in treating patients who do not respond to treatment with radioactive iodine or who have unresectable tumours, and also in those patients who present with medullary thyroid cancer.52–54
Furthermore, it has been reported that the compounds GDC-0879 and PLX4720 selectively inhibit the kinase activity of BRAFV600E in vitro, which results in the tumour cells reducing their proliferation. However, these have only proven effective in preclinical trials. On the other hand, it has been observed that patients who present with the BRAFV600E mutation may develop resistance to this type of compounds, since they increase the Raf-1 expression by activating Ras (generally over expressed) and thus the downstream signalling cascade towards ERK may continue being active. This event proves the need for a precise genotyping for each type of tumour, since it is necessary to ensure that the possible mutations have been detected in a few or several components of the signalling chain components which activate ERK, since, although one component is nullified, another may be activated and continue with the tumour proliferation process. That previously stated also proves that treatment should be include several compounds at the same time to ensure a better inhibitory outcome of the signalling cascade, and all the more so when tumour resistance to chemotherapy alone has been demonstrated and in the case of those who present with the mutation BRAFV600E, this type of combined treatment is the most recommended.53,54
The PI3K/Akt/mTOR signalling pathway is another appealing molecular target, due to its therapeutic benefit reported for different malignant tumours. The PI3K/Akt/mTOR pathway regulates critical cellular processes such as proliferation, apoptosis, the cellular cycle, metabolism and angiogenesis, whilst several preclinical and clinical studies support that the pharmacological inhibition of PI3K/Akt/mTOR represents a well tolerated and successful strategy in malignant tumour treatment, such as prostate, breast, colon, ovarian, lung and melanoma cancer.55 Apparently, the reduction in the activity of PI3K/Akt/mTOR is related to the induction of tumour radiosensibility effects,56 which are pro-apoptotic effects in tumour cells related to lower autophagia, and to the loss conservation in cancer cells.57 Regarding follicular type thyroid cancer, advanced or non differentiated anaplasia, resistant to radioactive iodine and therefore with a poor prognosis, for its response to convention treatment, several pre-clinical and clinical studies show that inhibition of PI3K/Akt/mTOR pathway may be a promising strategic target, whether with monotherapy or combined therapy, in this type of advanced thyroid cancer.58
ConclusionsMajor progress has been made during the last 30 years in the field of molecular biology aimed at discerning the mechanisms involved in the process of tumour transformation, primarily in studies intended to understand the intracellular systems which involve Ras/Raf/MEK/ERK kinase cascades. This signalling cascade controls cellular proliferation in normal cells and cancer cells. Moreover, they are important pathways in the development of resistance to the drugs used in chemotherapy.
Current studies have proven that the Ras/Raf/MEK/ERK signalling cascade induces malignant proliferation, since it stimulates cellular growth and simultaneously inhibits apoptosis (programmed cell death). Furthermore, the discovery that mutations in the BRAF gene may alone lead to cellular transformation and proliferation in several types of cancer, mainly thyroid cancer, has highlighted the need for further studies to be conducted on the control mechanisms of this type of genes and their products. This would include the development of effective chemical agents to nullify this type of protein and thus control the growth and development of cancer cells.59 That expressed above also underlines the need for greater study of tumour genotyping, due to their significant in molecular therapy usage. The description of mutations in BRAF may predict the sensitivity of B-Raf to several anti-cancer agents and inhibit the downstream to ERK and therefore inhibit growth and development of tumours. Genotyping of the tumour is also useful for predicting clinical behaviour of the tumour, to ensure correct administration of drugs and avoid confusion regarding the type of drugs to be applied when there is the possibility of resistance from tumours. Worldwide clinical research indicates that molecular therapy aimed against mutations in the BRAF may be promising. However, there is still much research to be carried out since each anti-cancer compound aimed at any component of the Ras/Raf/MEK/ERK chain must prove its clinical usage in patients with cancer. However, the application at the same time of several types of drugs for inhibiting the Ras/Raf/MEK/ERK chain and the combination with standard chemotherapy appears to be the most effective plan for thyroid cancer treatment. Notwithstanding, the risk of developing resistance is always latent, and the development of further and improved products for cancer control treatment is an absolute priority, especially if the development of these drugs led to more generalised usage and their availability in hospitals became standard, thus promoting wellbeing for the cancer patient.
Ethical disclosureProtection of people and animalsThe authors declare that for this research no experiments on humans or animals have been conducted.
Data confidentialityThe authors declare that the protocols of the centre of work have been adhered to regarding the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appears in this article.
Conflict of interestsThe authors have no conflicts of interest to declare.
Dr. Paul Mondragón Terán anbd Dr. José Gutiérrez Salinas would like to give thanks to the support from the “Programa de Investigación Científica y Tecnológica del ISSSTE” (Key E015). The authors thank the biologist Miguel Ángel Juárez Mancera for his support in reviewing the references. They would like to thank Mr. Sergio Hernandez Rodriguez for his support in editing the figures and to Miss. Cynthia Santiago Nicolas (Division de Investigation Biomedical, CAN 20 de November, ISSUE) for her help in secretarial tasks.
Please cite this article as: Mondragón-Terán P, López-Hernández LB, Gutiérrez-Salinas J, Suárez-Cuenca JA, Luna-Ceballos RI, Erazo Valle-Solís A. Mecanismos de señalización intracelular en cáncer de tiroides. Cir Cir. 2016;84:434–443.