





Introducción
Para comprender las alteraciones del equilibrio hidroelectrolítico y ácido-base, es decir, del "medio interno", así como sus graves repercusiones en la supervivencia de pacientes con peritonitis, han de definirse escuetamente unos conceptos y resumir unas alteraciones fisiopatológicas muy complejas, que explican el profundo trastorno, la mayoría de las veces irreversible, que se produce en la homeostasis de estos enfermos.
En 1911 Schottmüller1 emplea por primera vez el término de sepsis, definiéndola como una reacción sistémica a la llegada masiva de bacterias al torrente circulatorio. Sin embargo, hoy sabemos que la reacción sistémica puede producirse sin que las bacterias invadan el torrente circulatorio.
En efecto, toda agresión por agentes físicos, químicos o biológicos se caracteriza por producir una destrucción textural, más o menos extensa, dependiente de la intensidad del agente vulnerante. La respuesta del organismo no es otra que la de tratar de eliminar los tejidos desvitalizados, contaminados o no, sin que por ello se produzca una "enfermedad autoinmune".
La eliminación de tejidos desvitalizados, estén o no contaminados, se produce como consecuencia de una reacción inflamatoria sistémica (SIRS)2.
Desde el punto de vista clínico, la SIRS se caracteriza por alteraciones de la temperatura (> 38° C o < 36° C), taquicardia (> 90 lat/min), taquipnea (> 40 resp/min) o PaCO2 < 4,3 kPa y alteraciones leucocitarias: leucocitosis (> 12.000 leucocitos/µl) o leucopenia (< 4.000 leucocitos/µl).
Cuando la SIRS se acompaña o es debida a una infección hablamos de sepsis2, aceptando así la definición propuesta por Schottmüller1. Ahora bien, la infección no sólo puede ser exógena sino también endógena, tal y como ocurre en la pancreatitis aguda, en el shock hemorrágico, etc., que si bien en un principio son agresiones estériles, terminan en una sepsis, al producirse una alteración de la barrera mucosa intestinal (BMI), con la subsiguiente translocación bacteriana, que permite la llegada masiva de gérmenes de la flora intestinal al torrente sanguíneo. De estas alteraciones ya nos ocupamos en publicaciones anteriores3,4.
La sepsis se inicia, generalmente, por la activación del complejo macrófago-linfocitario (CML) y del sistema del complemento por endotoxinas, que no son más que lipopolisacáridos (LPS), constituyentes de la membrana celular de las bacterias gramnegativas. Su fracción más tóxica es el denominado lípido A (un fosfoglicolípido)5.
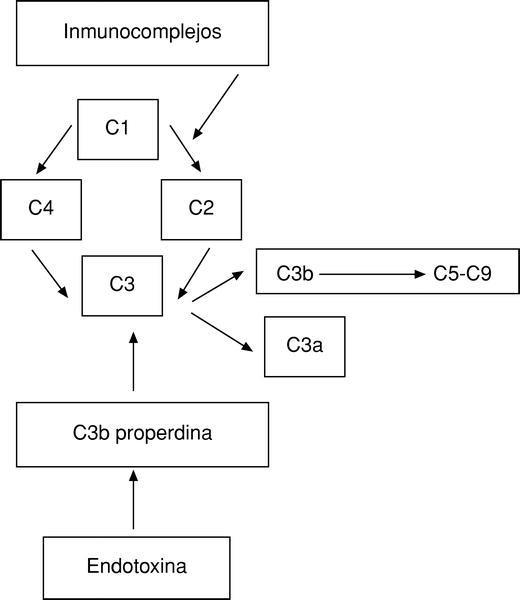
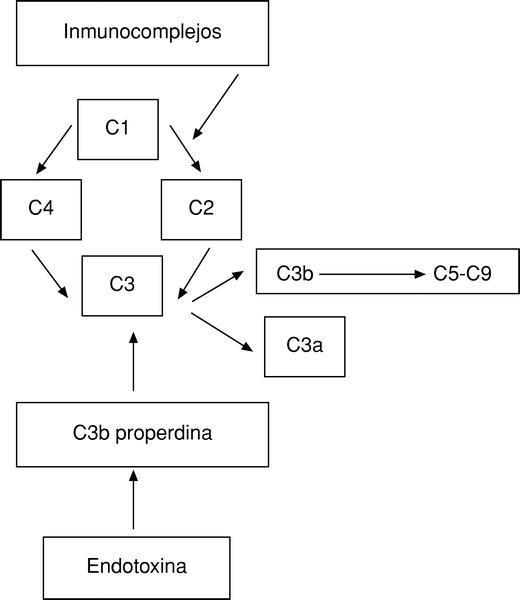
El sistema del complemento representa, junto con las inmunoglobulinas, el primer mecanismo defensivo humoral del organismo. Se puede activar por dos vías:
Directa. Por la acción de inmunocomplejos con anticuerpos IgG o IgM.
Indirecta. Merced a la estimulación del sistema de la properdina por la endotoxina.
Por ambas vías se activa el factor más importante del complemento, el C3, produciéndose a continuación una reacción en cascada6 (fig. 1).
La activación del complejo macrófago-linfocitario, y también de las células endoteliales, musculares lisas y granulocitos, origina la liberación de una serie de mediadores endógenos, que constituyen las defensas celulares de la respuesta inflamatoria. Su objetivo inicial no es otro que la eliminación de los agentes patógenos del organismo3,4.
Así, por ejemplo, el sistema microbicida de los PMN (granulocitos) está constituido por mieloperoxidasa, O2, OH, H2O2, hidrolasas lisosomales, lisozima, lactoferrina, defensina, etc.
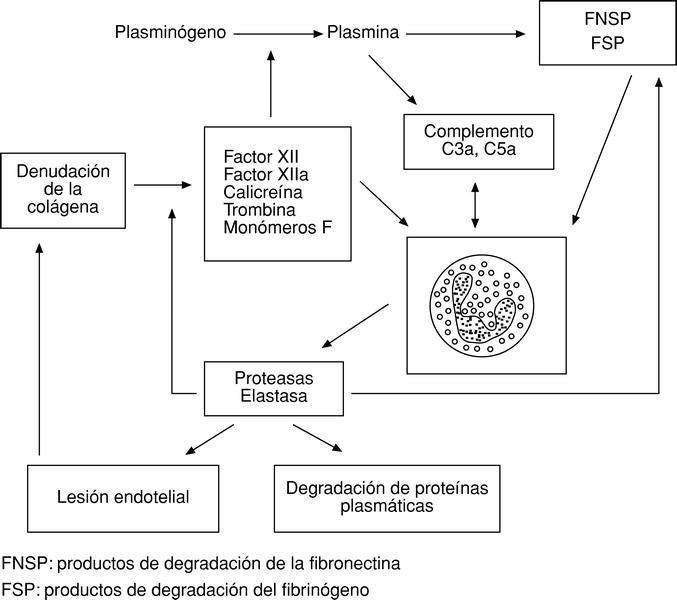
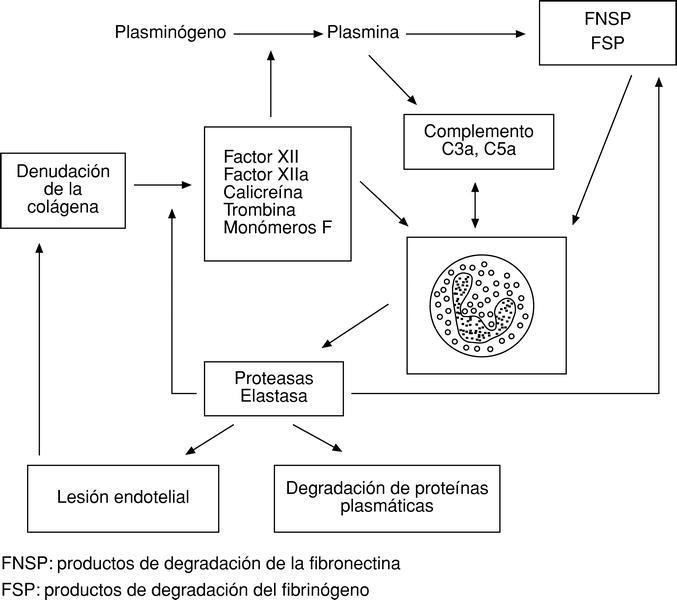
Existe una clara interdependencia entre los mecanismos defensivos humorales y celulares. Por poner un solo ejemplo, la denudación de la colágena por lesión endotelial provoca la activación de una serie de factores del sistema de la coagulación (también defensivo) que, actuando sobre el sistema del complemento y los granulocitos, incrementan la respuesta celular de los polimorfonucleares6 (fig. 2).
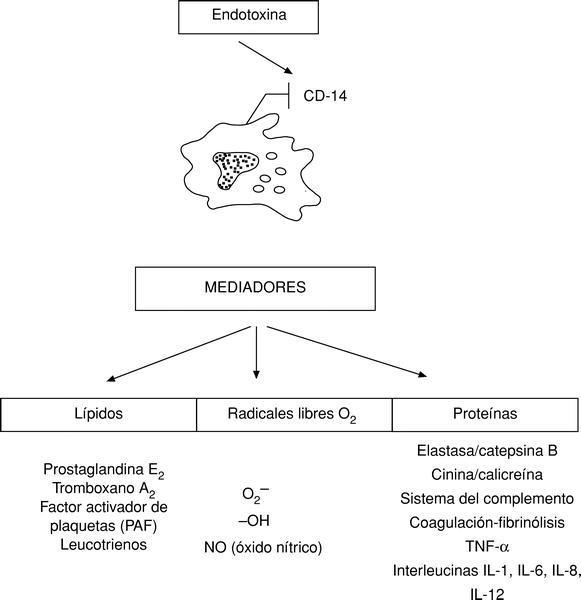
Sin embargo, la mayoría de los mediadores conocidos hasta la actualidad son producidos, liberados y activados por los macrófagos, distinguiéndose mediadores proteicos, radicales libres de O2 y lípidos (fig. 3).
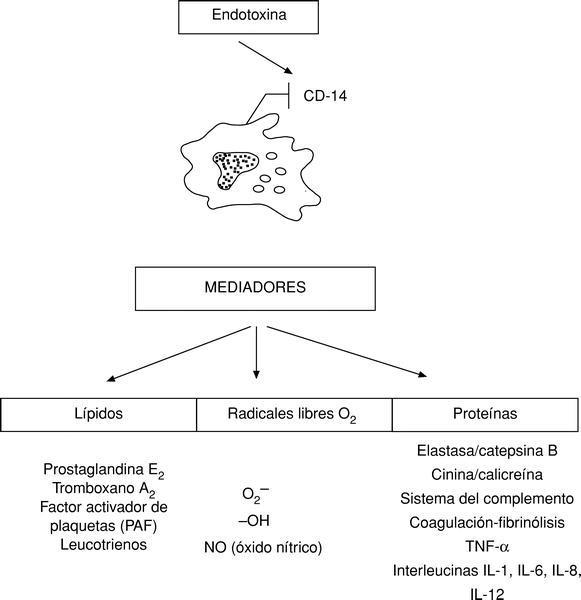
Todos estos mediadores endógenos o citocinas a concentraciones bajas tienen efectos beneficiosos para el organismo y constituyen la respuesta inflamatoria sistémica defensiva del mismo (SIRS). Así, por ejemplo, el TNF-* regula la actividad celular y la función de los leucocitos y células endoteliales; sin embargo, este mismo factor a altas concentraciones se fija en los receptores p55 y p75, existentes prácticamente en todas las células produciendo su muerte, por apoptosis, o bien su activación originándose más TNF-* que potencia a las endotoxinas y termina por desencadenar una enfermedad autoinmune, un autocanibalismo, un horror autotoxicus4,7.
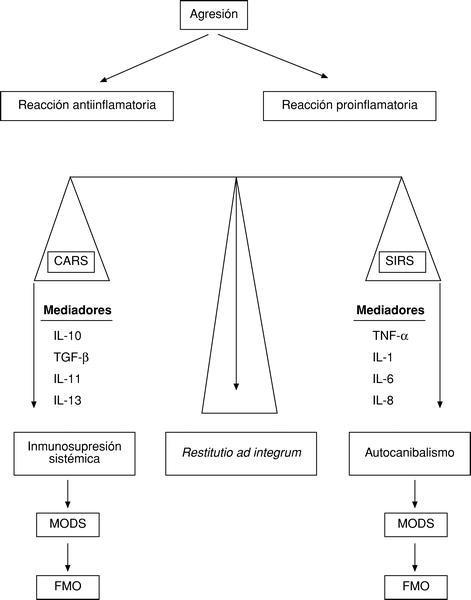
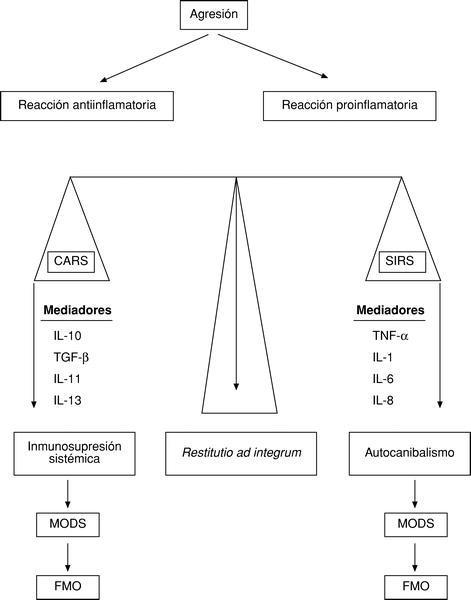
Para que esto no suceda, el organismo es capaz de desarrollar una reacción antiinflamatoria, denominada por Bone CARS (Compensatory antiinflammatory response syndrom), que propicia una inmunosupresión. Así pues, ante toda agresión se activaría no sólo una reacción proinflamatoria (SIRS), sino también una reacción antiinflamatoria (CARS). El equilibrio entre ambas permitiría una restitutio ad integrum. El predominio de la SIRS daría origen al autocanibalismo, un síndrome de disfunción multiorgánica (MODS) y, por ende, al fallo multiorgánico (FMO); pero si predomina la CARS, se producirá una inmunosupresión sistémica que propiciará la sepsis, el MODS y el FMO9 (fig. 4).
Pues bien, dentro de la sepsis (entendida como SIRS más infección) se distinguen, desde el punto de vista clínico, dos es-
tadios2.
Sepsis grave
Cuando aparece hipoperfusión (acidosis metabólica, oliguria y/o graves alteraciones de la conciencia), o bien hipotensión (presión arterial sistólica < 90 mmHg o disminución de la habitual de más de 40 mmHg). Esta sepsis grave se conoce también bajo el nombre de síndrome de disfunción multiorgánica (MODS).
Shock séptico
Caracterizado por hipotensión mantenida a pesar de aportar un suficiente volumen de líquidos y los correspondientes fármacos inotrópicos y vasopresores para mantener la función cardiovascular. El shock séptico se caracteriza por cursar con un FMO, y fallecen el 82% de los pacientes.
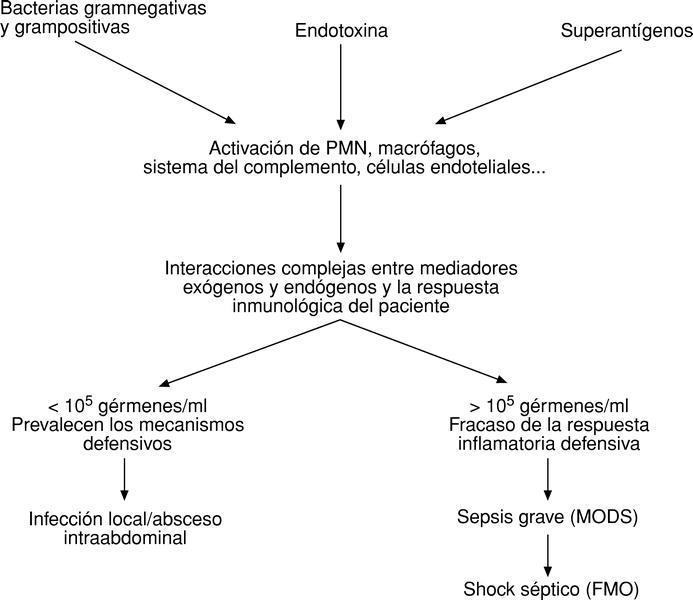
Ahora bien, las endotoxinas de bacterias gramnegativas no son las únicas desencadenantes de la sepsis. En la actualidad se sabe que las exotoxinas de bacterias gramnegativas, como la hemolisina producida por E. coli, la *-toxina de Staphylococcus aureus o los superantígenos de bacterias grampositivas, también pueden ser causantes de una sepsis10,11.
La peritonitis no es más que una sepsis, localizada en la cavidad abdominal, pero que puede y suele evolucionar hacia una sepsis generalizada, que afecta a todos los órganos y sistemas del organismo, originando un shock séptico, de fatal desenlace.
Peritonitis
La peritonitis, inicialmente, es una respuesta inflamatoria del peritoneo, tanto parietal como visceral, ante la agresión que virus, bacterias o agentes químicos producen al invadir la cavidad peritoneal, originando la activación local de los sistemas defensivos humorales y celulares.
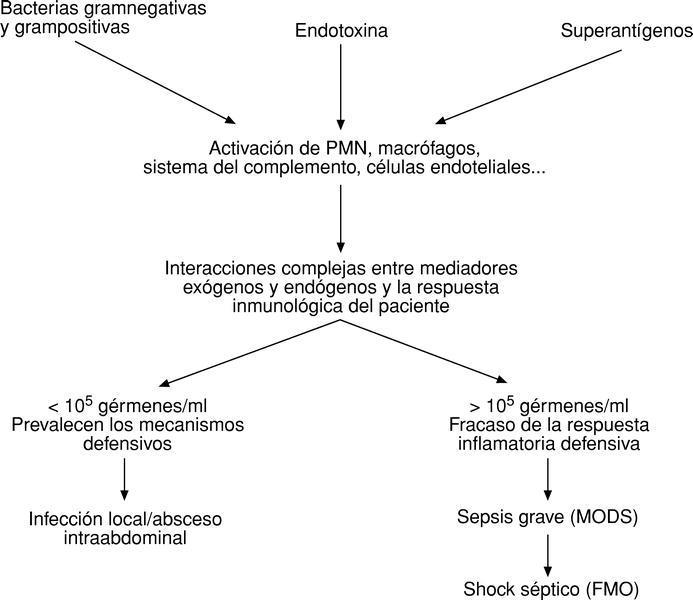
En un principio, el peritoneo mantiene aislada la cavidad peritoneal del torrente circulatorio y así, puede ocurrir que los sistemas defensivos sean capaces de localizar la infección (peritonitis localizada, abscesos intraabdominales). Hoy se cree que el peritoneo es capaz de impedir la llegada de bacterias, así como de los mediadores activados al torrente circulatorio, siempre y cuando la concentración de gérmenes por ml de líquido peritoneal sea menor de 105; si la concentración en líquido peritoneal supera los 105 gérmenes/ml, éstos, sus productos tóxicos y los mediadores activados alcanzan, a través de los linfáticos submesoteliales (el peritoneo es un mesotelio de 2 m2), el torrente circulatorio provocando una sepsis grave con MODS o un shock séptico y FMO12,13 (fig. 5).
En la actualidad, el peritoneo no puede considerarse como una membrana con funciones exclusivamente defensivas, sino como una membrana metabólica muy activa, que con la ayuda de factores humorales y celulares defensivos, transforma su función protectora en otra agresiva, autotóxica y autodestructiva. La activación de PMN, macrófagos, células mesoteliales pluripotentes, etc., tan numerosas en el peritoneo, tiene, evidentemente, una función defensiva, pero pueden desencadenar una enfermedad sistémica autoinmune, autotóxica, un catabolismo séptico, si existen:
Suficientes focos infecciosos activos.
Alteraciones de la función transportadora por disfunciones de la permeabilidad de membrana, que permiten el paso masivo de noxas locales al torrente circulatorio12.
El resultado final de todos estos mecanismos y reacciones no es otro que la aparición de una alteración de la permeabilidad de todas las barreras de los distintos órganos, incluida la hematoencefálica12,14.
Podemos resumir afirmando que todos los agentes agresivos, especialmente las endotoxinas, determinan la liberación de sustancias proteolíticas, vasoactivas y radicales libres de O2, bien por vía directa, activando principios no activos subendoteliales o bien indirectamente, estimulando las funciones de los PMN y macrófagos del peritoneo, hígado y mucosa intestinal. Es necesario señalar que prácticamente todos los mediadores, excepción hecha de las toxinas de los quemados, se producen y activan en el mismo territorio esplácnico.
Todos estos mediadores además de aumentar la permeabilidad de las barreras de los distintos órganos, originan un trastorno de la microcirculación: la vasomotrocidad disminuye o desaparece, se produce una agregación leucoplaquetaria (leucotaxis) y un edema de las células endoteliales15.
Precisamente estas modificaciones de la permeabilidad y de la microcirculación son las responsables de las alteraciones hidroelectrolíticas y del equilibrio ácido-base que se originan en el paciente con peritonitis.
El íleo paralítico. Alteración del equilibrio hidroelectrolítico
Es bien conocido que en toda peritonitis se produce un íleo paralítico reflejo que, en principio, tiene por finalidad dejar en reposo el tracto gastrointestinal, limitando así la difusión del proceso peritonítico16.
Sin embargo, el íleo paralítico da origen a importantes alteraciones de la mucosa intestinal. En efecto, la capacidad de reabsorción del intestino en condicionantes normales alcanza los 6-8 l/24 h. Se reabsorben principalmente soluciones isotónicas hidroelectrolíticas, 200-300 g de albúmina (secretada por las glándulas intestinales), y varios litros de gas que se producen como consecuencia de la fermentación de sustratos, del metabolismo bacteriano y del aire deglutido.
Esta falta de reabsorción determina una distensión de las paredes intestinales, favorecida por la atonía de sus células musculares lisas debida, fundamentalmente, al déficit de potasio.
Por otro lado, el estancamiento del tránsito intestinal provoca una intensa proliferación de las enterobacterias, especialmente de los colibacilos. Aun cuando la mucosa intestinal se encuentra protegida por la gran cantidad de inmunoglobulina A existente en el moco llega un momento en que ésta es insuficiente y las enterobacterias, posiblemente a través del AMP cíclico (AMPc), estimulan a la mucosa intestinal produciéndose un incremento de la secreción. Pero es que, además, las células de la mucosa activan el sistema de las prostaglandinas (en estas células se segregan y almacenan gran cantidad de PG), que incrementan el flujo sanguíneo de la misma, lo que favorece por un lado, la hipersecreción y, por otro, la vasodilatación capilar con aumento de la permeabilidad y subsiguiente edema de la pared intestinal17.
El aumento de la permeabilidad de la pared intestinal permite el paso de fluidos a la cavidad abdominal, en forma de exudado o trasudado, originando un secuestro de líquidos en el llamado tercer espacio (asas intestinales, cavidad peritoneal), que disminuyendo el volumen circulante, favorece la aparición de un shock hipovolémico18.
Si se recuerda la composición del jugo gastrointestinal (tabla 1), se comprenderá la importancia del trastorno electrolítico.

En el íleo paralítico la falta de reabsorción y la hipersecreción determinan una depleción importante de sodio y también, aunque menor, de cloro; pérdidas que se acentúan aún más si se producen vómitos reflejos.
Pero además del secuestro de Na+ y Cl ocasionado por la falta de reabsorción y aumento de secreción de la mucosa gastrointestinal, se produce una pérdida aún mayor de estos electrólitos como consecuencia de los trastornos de la microcirculación que acompañan a todo íleo. Estas alteraciones determinan un aumento de permeabilidad capilar, edema generalizado de las paredes intestinales con secuestro de plasma, que termina drenándose en cavidad peritoneal, en forma de ascitis.
Por otro lado, la hipoxia celular, que los trastornos microcirculatorios entrañan determina una alteración de la bomba celular sodio/potasio, con entrada de Na+ y salida del K+ al espacio intercelular.
Todos estos mecanismos explican que en el íleo paralítico se produzcan elevadísimas pérdidas de Na+, instaurándose una grave acidosis metabólica, favorecida por la hipoxia y las precoces alteraciones de la función respiratoria.
Con el potasio sucede lo mismo; se originan grandes pérdidas por la falta de reabsorción e hipersecreción. El potasio se elimina por los vómitos o bien queda secuestrado en la luz intestinal. En el íleo paralítico se han encontrado, en el contenido intestinal, hasta 10-12 mEq/l de potasio18. Se produce, pues, muy precozmente, una hiponatremia, que puede estar enmascarada por el balance negativo de agua, y una hipopotasemia, que al no permitir la llegada de este ion a las células de la musculatura lisa intestinal, aumenta aún más la atonía y distensión paralítica de las asas, creándose así un auténtico círculo vicioso. No hace falta decir que la hipopotasemia puede producir trastornos del ritmo e incluso, la parada del corazón.
Kurek et al19 demostraron, en 1995, una estrecha relación entre la homeostasis del potasio y el metabolismo energético de los hidratos de carbono. Así, una hipopotasemia reduce la oxidación de la glucosa por la vía de la fosfatopentosa, lo que disminuye la concentración de adenilnucleótidos y aumenta la permeabilidad de membrana.
La pérdida hídrica se correlaciona con la de los iones de sodio y potasio. La acumulación de agua en el tercer espacio (luz intestinal, paredes edematosas y cavidad peritoneal: ascitis), provoca una auténtica exicosis, ya que se acumula en dicho espacio una cantidad de agua que representa más del 10% del peso corporal. La gravedad de la hipovolemia provocada guarda una relación directa con la taquicardia, la oliguria y la hemoconcentración.
La presión intraabdominal medida a través de la vejiga urinaria oscila, en condiciones normales, alrededor de 10 mmHg. Un incremento igual o mayor a 15 mmHg indica la existencia de un síndrome compartimental del abdomen20.
El aumento de la presión intraabdominal, propiciado por el íleo paralítico y la ascitis, determina un síndrome compartimental, que afecta muy precozmente a la función renal, pues la compresión colapsa las arterias y venas renales, produciendo una disminución del filtrado glomerular y una uremia.
Pero, además, la compresión de la vena cava inferior dificulta el retorno de la sangre de las extremidades inferiores, con lo que aumenta el secuestro sanguíneo. Disminuye así, aún más, el retorno venoso al corazón, descendiendo drásticamente el gasto cardíaco. Se propicia la aparición, por estasis, de trombosis venosas profundas con sus complicaciones (embolismo pulmonar y síndrome postrombótico).
El aumento de la presión intraabdominal determina una elevación del diafragma, que reduce la capacidad ventilatoria, hecho que favorece la instauración de un síndrome de insuficiencia respiratoria aguda (ADRS).
El síndrome de insuficiencia respiratoria aguda y las alteraciones del equilibrio ácido-base
Es bien conocido que mediadores producidos durante la SIRS y el íleo paralítico, como la prostaglandina E2 y el leucotrieno L4, inducen precozmente un ARDS7.
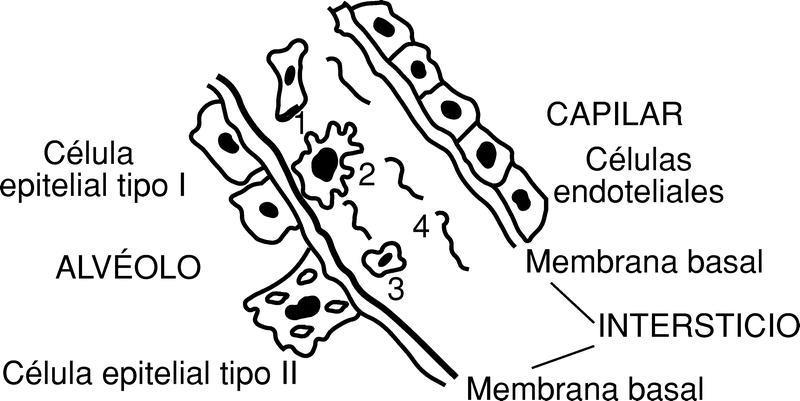
En efecto, el intercambio gaseoso en el pulmón se realiza a través de la barrera alveolocapilar (fig. 6).
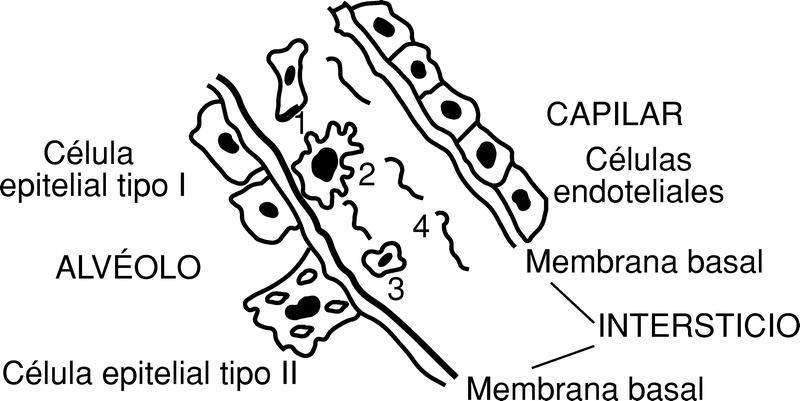
Dicha barrera está constituida por las células endoteliales del capilar, el intersticio y la pared alveolar, constituida por células epiteliales: tipo I (células planas) y tipo II (células cuboideas con microvillis que secretan el surfactante y que son, además, las progenitoras de las células epiteliales del alvéolo).
El intersticio alveolar comprende las membranas basales (lámina elástica interna). El espacio intersticial está ocupado por células mesenquimales, fundamentalmente macrófagos, y algunos linfocitos, con evidentes propiedades inmunológicas (no hay que olvidar que el pulmón es el segundo filtro depurador más importante del organismo). Junto con las células mesenquimales el intersticio cuenta con una estroma constituida fundamentalmente por fibras colágenas tipo I y III, elastina, fibronectina y proteoglicanos.
En la sepsis, como consecuencia de la liberación de mediadores tales como PGE2 y L4, así como de otras muchas citocinas y radicales libres de O2, se produce, al intentar ser filtradas y depuradas por el pulmón, una muy precoz alteración del intersticio alveolar. En efecto, las células mesenquimales se activan, liberando más mediadores y el intersticio aumenta de grosor, al ser ocupado por fluidos plasmáticos ricos en proteínas, de gran peso molecular. Al mismo tiempo, se produce una agregación leucoplaquetaria en los capilares sanguíneos; sin embargo, las células endoteliales microscópicamente no están alteradas, pero sí las células epiteliales de tipo I. Este hecho produce una dificultad en el intercambio gaseoso, por un trastorno de la permeabilidad de la barrera alveolocapilar. A las 24 h de la agresión, el intersticio aparece ocupado por neutrófilos, macrófagos y linfocitos, así como por plaquetas y fibrina. Se destruyen las células epiteliales tipo I y aparecen soluciones de continuidad en la pared alveolar, que se comunica directamente con el espacio intersticial. Al desaparecer la barrera alveolar el agua y los solutos del capilar pasan al alvéolo; 48 h más tarde, se produce una coagulación intravascular, ocluyéndose los capilares no sólo por fibrina sino especialmente por agregados leucoplaquetarios (leucotaxis); las células endoteliales se destruyen. La barrera alveolocapilar desaparece y, por consiguiente, el intercambio gaseoso, produciéndose una hipoxia y subsiguiente acidosis metabólica21.
La gravedad del ARDS es obvia, pues fallecen entre el 70-80% de los pacientes.
Conclusión
La peritonitis generalizada es una sepsis grave. Como consecuencia de endotoxinas y exotoxinas se produce una hiperreacción inflamatoria defensiva que, sobrepasando al CARS, origina una enfermedad autoinmune, un autocanibalismo séptico.
En efecto, la disfunción multiorgánica reconoce su origen en dos hechos fisiopatológicos esenciales: el aumento de la permeabilidad de todas las barreras del organismo, incluida la hematoencefálica y los trastornos de la microcirculación que conducen a una hipoperfusión textural.
Estas alteraciones mantienen y agravan el íleo paralítico (inicialmente un mecanismo defensivo) y originan su grave ARDS, lo que unido a la hipovolemia provoca un shock séptico cuya mortalidad alcanza hasta más del 82% de los casos7,14. Además, de los supervivientes, un 26% no alcanzan más de un año de vida, falleciendo por causas no sépticas22.
Implicaciones terapéuticas
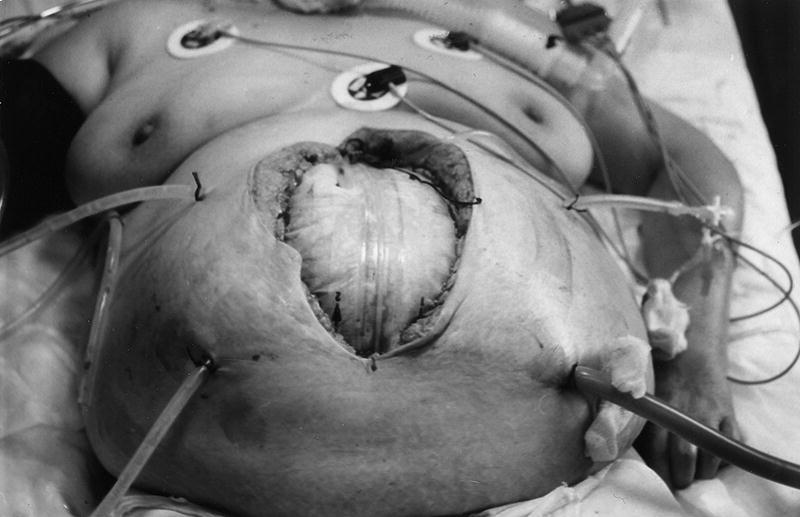
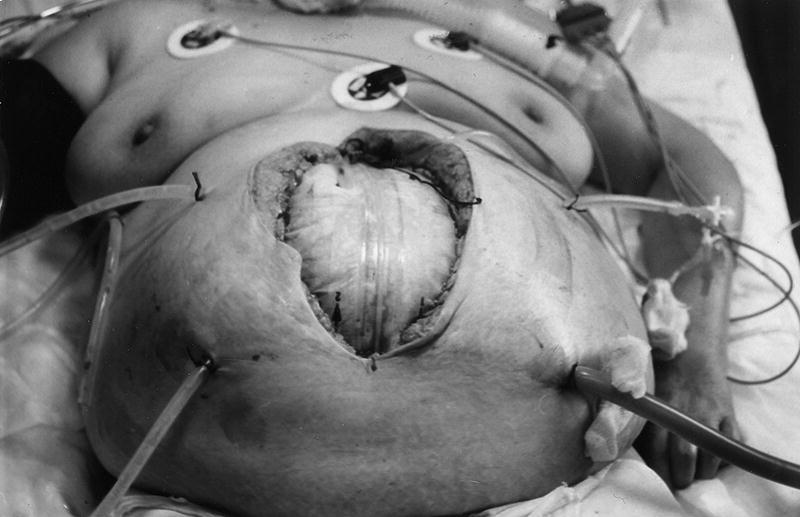
1. Si se exceptúan las raras formas de peritonitis primaria (peritonitis por neumococo del niño, la llamada "peritonitis espontánea del cirrótico, pelvicoperitonitis, etc.), subsidiarias de un tratamiento conservador con antibióticos, el tratamiento de las peritonitis secundarias (90%) es siempre quirúrgico. Se ha de conseguir la erradicación total del foco infeccioso, pues es la única forma de romper el círculo vicioso de la sepsis peritoneal. De ahí que deba realizarse una cirugía agresiva y eficaz. En algunos casos (pancreatitis, etc.), esto no es posible, de ahí que haya que combinar una cirugía resectiva primaria con relaparotomías planificadas, a demanda o laparotomías abiertas (fig. 7). Todas estas reintervenciones incrementan la respuesta inflamatoria y con ello la mortalidad. Por esto, en la actualidad, se vuelve a recomendar la irrigación o lavado continuo peritoneal14, que nosotros defendimos ya en 198223.
2. El íleo paralítico, como hemos visto, desempeña un papel importantísimo en la fisiopatología de la peritonitis. Por tanto, debe prevenirse mediante la administración de simpaticolíticos y parasimpaticomiméticos, o bien mediante la anestesia mesentérica local (AML), como nosotros propusimos ya en 198824. Además, la AML mejora significativamente el gasto cardíaco25.
Si el íleo está establecido, la terapia de elección es la anestesia epidural continua entre las dermatomas T5-L1.
La aspiración nasogástrica y la descompresión colonoscópica completan las medidas básicas y fundamentales de fluidoterapia, encaminadas a restituir la volemia y el equilibrio hidroelectrolítico y ácido-base.
3. El ARDS ha de tratarse precozmente, pues es la única manera de mejorar el aporte de oxígeno a los órganos y tejidos, combatiéndose así la acidosis metabólica.
La oxigenoterapia y la monitorización respiratoria mejoran la función residual del pulmón, la oxigenación arterial y la oferta de O2 a los tejidos. Si el enfermo respira espontáneamente utilizaremos la presión positiva continua en vías aéreas (CPAP); si está conectado a un respirador la presión positiva al final de la espiración (PEEP). En todos los casos debe vigilarse la presión venosa central, para mantener el balance hidroelectrolítico ligeramente negativo.
Sustancias vasoactivas, tales como la dopamina y la adrenalina, están indicadas por sus efectos vasoconstrictores y, por ende, ahorradores de volumen. Antibioterapia y corticoterapia, a altas dosis, han de administrarse precozmente.
4. En el momento actual, ningún estudio bien diseñado ha podido demostrar la efectividad de ningún tratamiento con antimediadores, por lo que éstos no pueden ser recomendados para el tratamiento de la sepsis peritoneal26.
Correspondencia: Dr. R. Vara Thorbeck. Cátedra de Patología y Clínica Quirúrgicas. Hospital Clínico San Cecilio. Avda. Doctor Oloriz, s/n. 18012 Granada.

