




El cáncer de mama (CM) es el tumor maligno más frecuente en la mujer como resultado de una combinación de factores físicos, químicos, biológicos y genéticos. La mayoría de los CM son esporádicos, menos del 10% presentan carácter familiar debido fundamentalmente a alteraciones en los genes BRCA1 y BRCA2, y tan solo un pequeño número de casos hereditarios pueden ser atribuidos a mutaciones en otros genes como el p53 en el síndrome de Li Fraumeni, el gen PTEN en el síndrome de Cowden, los genes MSH2 y MLH1 en el síndrome de Muir-Torre, el gen ATM en la ataxia-teleangiectasia y el gen STK11 en el síndrome de Peutz-Jeghers (SPJ)1.
El SPJ constituye una entidad infrecuente caracterizada por la asociación de pólipos hamartomatosos en el tubo digestivo, lesiones hiperpigmentadas mucocutáneas y un incremento en el riesgo de desarrollar neoplasias digestivas y extradigestivas. Su patrón hereditario es autosómico dominante (AD) de penetración variable y en un alto porcentaje (80-94%) se asocia a mutaciones en el locus específico 19p13.3 del gen LKB1/STK112,3.
El SPJ, definido por Bruwer et al.4 en 1954, se engloba dentro del grupo de las poliposis hamartomatosas junto a la poliposis juvenil, el síndrome de la poliposis mixta hereditaria, el síndrome de Cowden y el síndrome de Ruvalcava-Myrhe-Smith, con los que comparte un riesgo elevado de cáncer colorrectal5. Existe, además, en el SPJ una alta probabilidad de neoplasias en el resto del aparato digestivo, y otras localizaciones en edad temprana, siendo las más frecuentes el cáncer de mama (54%), de ovario (21%), de cérvix (19%) y de pulmón (15%)5,6.
Este raro síndrome (1/200.000 nacidos vivos), que afecta por igual a ambos sexos, habitualmente se diagnostica durante la infancia a partir de los antecedentes familiares, por la presencia de manchas melánicas típicas, o a través de los problemas derivados del crecimiento de los pólipos intestinales que ocasionan cuadros de invaginación, obstrucción o hemorragia.
Aunque la pigmentación puede aparecer a cualquier edad, es más frecuente durante la lactancia y primera infancia, con depósitos melánicos en mucosa labial (95%) u oral (83%), siendo menos frecuente en manos, pies y localización periorificial. La mayoría de las manifestaciones clínicas se producen a nivel gastrointestinal, aunque la secuencia hamartoma-adenoma-carcinoma es controvertida para algunos autores7 y se conoce como «landscape effect», donde las alteraciones, sobre todo de la lámina propia, pueden conducir a cánceres epiteliales8. En otras ocasiones la primera manifestación es la aparición de una neoplasia en edad temprana debido a la acumulación de riesgos.
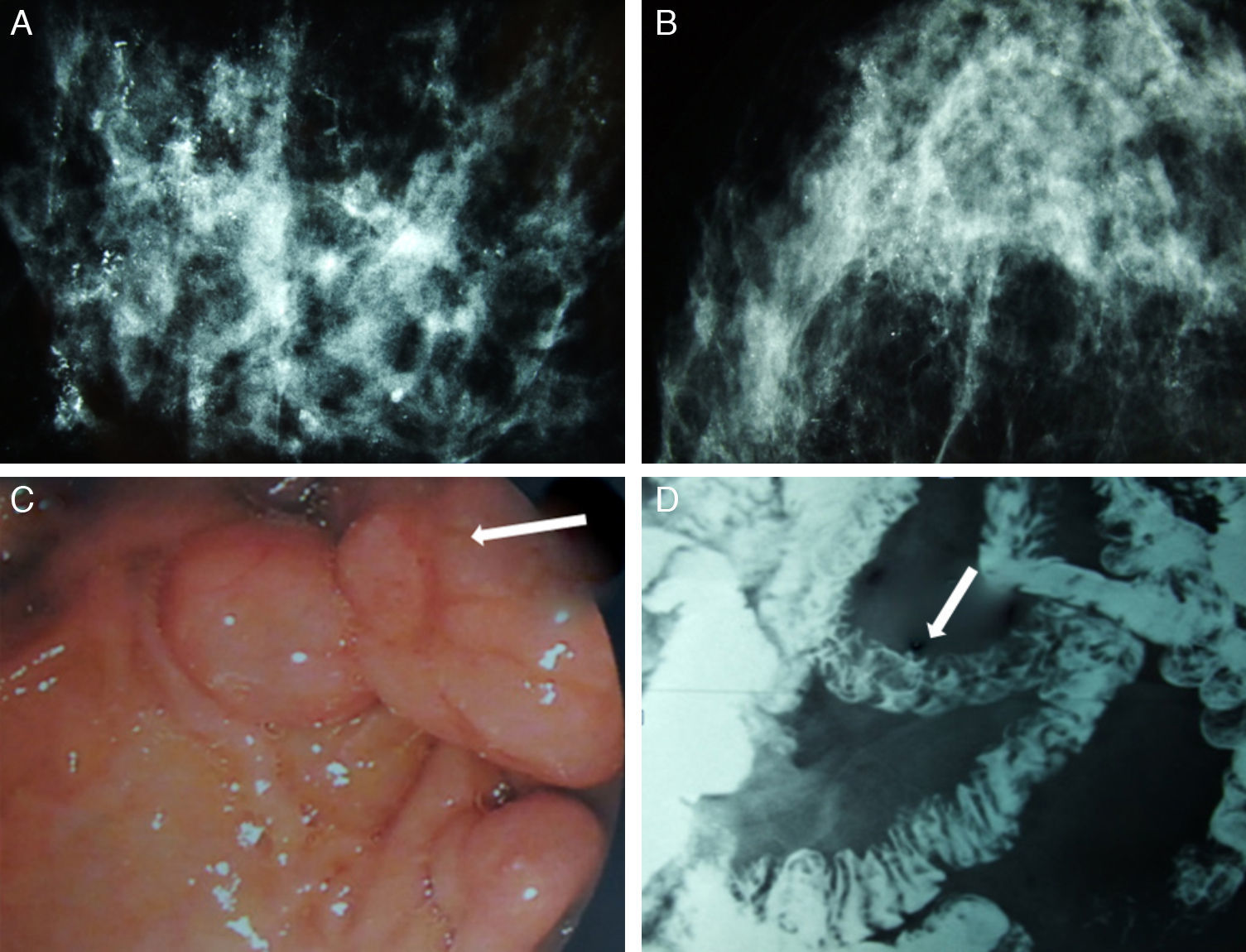
Presentamos el caso de una paciente afecta de CM en la que se descubre el carácter hereditario de este de manera casual. Se trata de una mujer caucásica de 51 años con antecedentes de anemia crónica, en tratamiento con hierro oral, y colecistectomizada. No relató antecedentes familiares de cáncer de mama ni alteraciones genéticas. Fue diagnosticada a la edad de 50 años de carcinoma bilateral de mama tras el hallazgo mamográfico de microcalcificaciones muy sospechosas, confirmadas mediante resonancia magnética (RM) (fig. 1). Se realizó mastectomía bilateral y estudio de extensión axilar mediante biopsia selectiva de ganglio centinela, con resultado negativo por lo que se procedió a la reconstrucción inmediata con prótesis/expansor tipo Becker. El examen anatomopatológico demostró que se trataba de un carcinoma intraductal de alto grado (índice de Van Nuys > 8) en ambas mamas.
 Microcalcificaciones difusas en la mama derecha. B) Microcalcificaciones difusas en la mama izquierda. C) La endoscopia digestiva alta revela pólipos hamartomatosos en la tercera porción duodenal (flecha blanca). D) Tránsito gastrointestinal donde se observan defectos de repleción a nivel de yeyuno proximal que corresponden a formaciones polipoideas (flecha blanca).")
Composición que recoge imágenes de la mamografía, tránsito baritado y endoscopia digestiva. A) Microcalcificaciones difusas en la mama derecha. B) Microcalcificaciones difusas en la mama izquierda. C) La endoscopia digestiva alta revela pólipos hamartomatosos en la tercera porción duodenal (flecha blanca). D) Tránsito gastrointestinal donde se observan defectos de repleción a nivel de yeyuno proximal que corresponden a formaciones polipoideas (flecha blanca).
La paciente acudió a Urgencias por molestias en hipocondrio derecho y náuseas, sin fiebre asociada, de escaso tiempo de evolución. En el análisis sanguíneo destacaba una discreta hipertransaminasemia, por lo que se completó el estudio con ecografía y colangio-RM siendo diagnosticada de coledocolitiasis. Durante la realización de la CPRE se evidenciaron numerosos pólipos en duodeno así como un cálculo coledociano que se extrajo. Mediante endoscopia digestiva alta (EDA) se extirparon varios pólipos gástricos y duodenales (fig. 1). La presencia de manchas melánicas peribucales y la confirmación histológica de pólipos hamartomatosos establecieron el diagnóstico de sPJ9.
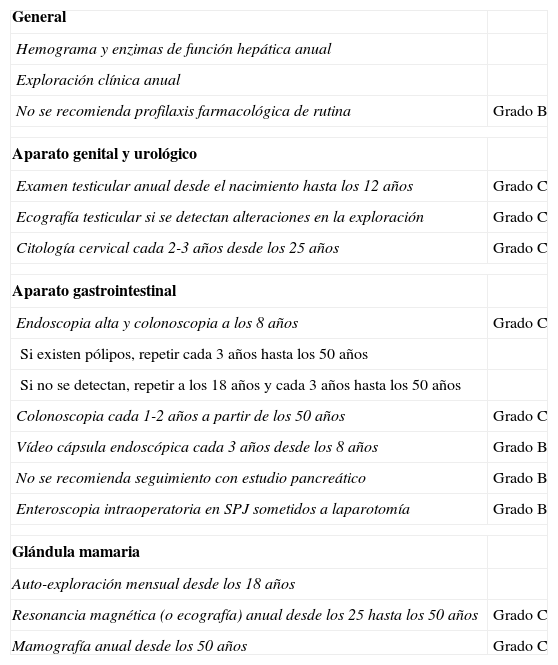
Todos los pacientes diagnosticados de SPJ deben ser controlados de por vida y remitidos a consejo genético de acuerdo con las recomendaciones propuestas por un Comité de Expertos Europeos reunidos en Mallorca en 2007 y que se recogen en un documento de consenso (guía clínica) con un nivel de evidencia B o C9. El objetivo es doble: 1) detectar la presencia de pólipos con un tamaño adecuado para su extirpación endoscópica, con lo que se evitan posibles complicaciones y 2) diagnosticar precozmente el cáncer.
Dichas recomendaciones, recogidas en la tabla 1, exigen un seguimiento estrecho y multidisplinar con exploraciones analíticas y de los aparatos digestivo y genitourinario así como exámenes mamarios seriados en una edad inferior a la que habitualmente se realizan. En la actualidad nuestra paciente sigue sometida a estos controles y pendiente del estudio genético que podrá definir el intervalo recomendable para el cribado y vigilancia dentro de su misma familia, evitando así futuras neoplasias.
Recomendaciones para la realización de exploraciones y el seguimiento en pacientes diagnosticados de síndrome de Peutz-Jeghers. Grados de recomendación
| General | |
| Hemograma y enzimas de función hepática anual | |
| Exploración clínica anual | |
| No se recomienda profilaxis farmacológica de rutina | Grado B |
| Aparato genital y urológico | |
| Examen testicular anual desde el nacimiento hasta los 12 años | Grado C |
| Ecografía testicular si se detectan alteraciones en la exploración | Grado C |
| Citología cervical cada 2-3 años desde los 25 años | Grado C |
| Aparato gastrointestinal | |
| Endoscopia alta y colonoscopia a los 8 años | Grado C |
| Si existen pólipos, repetir cada 3 años hasta los 50 años | |
| Si no se detectan, repetir a los 18 años y cada 3 años hasta los 50 años | |
| Colonoscopia cada 1-2 años a partir de los 50 años | Grado C |
| Vídeo cápsula endoscópica cada 3 años desde los 8 años | Grado B |
| No se recomienda seguimiento con estudio pancreático | Grado B |
| Enteroscopia intraoperatoria en SPJ sometidos a laparotomía | Grado B |
| Glándula mamaria | |
| Auto-exploración mensual desde los 18 años | |
| Resonancia magnética (o ecografía) anual desde los 25 hasta los 50 años | Grado C |
| Mamografía anual desde los 50 años | Grado C |
Tomada de Beggs AD et al.9.
SPJ: síndrome de Peutz-Jeghers.

