





Introducción
El carcinoma suprarrenal es un tumor raro pero devastador, fundamentalmente debido a que en la mayoría de los casos el tumor se encuentra en estadios avanzados en el momento del diagnóstico. Esto suele ser la consecuencia de la gran tendencia que tiene a invadir las estructuras vasculares, lo que origina una metastatización precoz, así como de la dificultad diagnóstica que, debido a su muy profunda situación, presentan las glándulas suprarrenales, lo que hace que cuando el tumor es palpable se encuentra ya avanzado. En tercer lugar, porque suele ser un tumor olvidado por los clínicos, que sólo piensan en él cuando se asocia a un síndrome hiperfuncional; sin embargo, la mayoría son no funcionantes y, en otras ocasiones, su forma clínica de expresión funcional queda encubierta como ocurre, por ejemplo, en los tumores con hipersecreción androgénica en el varón. En cuarto lugar, se ha mantenido la idea de que es un tumor incurable, lo que supone un gran error puesto que diagnosticado en estadios precoces es potencialmente curable, y diagnosticado en estadios más avanzados puede, bajo un tratamiento agresivo, ofrecer un grado no desdeñable de remisión. Por último, las modernas técnicas de imagen permiten realizar diagnósticos mucho más precoces, en momentos en los que el tumor es aún asintomático.
Epidemiología y etiología
El carcinoma suprarrenal presenta una incidencia global entre el 0,5-2/1.000.000 habitantes, representando el 0,02% de los cánceres. En incidentalomas adrenales, las cifras oscilan alrededor del 2-3%1, aunque se han publicado series con cifras tan elevadas como 13%2. Afecta a ambos sexos, con una discreta preponderancia femenina, especialmente hacia el final de la tercera y cuarta décadas, aunque puede presentarse en edades extremas. Los tumores no funcionantes suelen ocurrir entre la cuarta y séptima década, con un predominio masculino, al contrario que los hiperfuncionales que, expresados en forma de hipercortisolismo y/o virilización, suelen ser más frecuentes en el sexo femenino. Aproximadamente el 50-60% de los casos presentan clara evidencia de diseminación locorregional o a distancia en el momento del diagnóstico.
Desde el punto de vista etiológico puede afirmarse que ésta es desconocida, no habiéndose identificado factores predisponentes. De todos modos, es intrigante la posible relación existente entre el desarrollo de carcinoma suprarrenal en glándulas que han sido crónicamente estimuladas con ACTH. En ciertas cepas de ratón se ha descrito el paso eventual de hiperplasia a carcinoma, y existen algunas comunicaciones de carcinomas en pacientes con síndrome adrenogenital no tratado, enfermedad de Cushing hipófiso-dependiente e hiperplasia macronodular, aunque la significación de estas comunicaciones en términos de poder aceptar una relación causal directa no está nada clara.
Anatomía patológica
La anatomía patológica del carcinoma adrenal debe ser considerada en términos de su aspecto macro y microscópico y en su inusual origen desde "células adrenales en reposo".
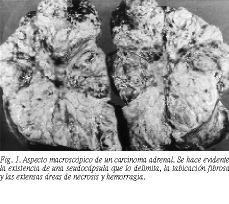
Macroscópicamente, es un tumor muy vascularizado. Su superficie de corte es rosácea o rojiza, blanda, al contrario que los adenomas que suelen presentar una consistencia firme, y con zonas más o menos grandes de necrosis y/o hemorragia (fig. 1). El carcinoma adrenal presenta una gran proclividad por invadir las estructuras adyacentes, así como los tejidos blandos del espacio retroperitoneal.

Microscópicamente puede ser dividido en diferenciado e indiferenciado o pleomórfico. Las formas diferenciadas están compuestas por células que se asemejan a las células normales de la corteza adrenal. Las células son poligonales, de apariencia monótona, y en general ordenadas en capas o en trabéculas, siendo menos frecuente la disposición en nidos o cordones. Las células contienen abundantes figuras de mitosis, y a menudo presentas focos de necrosis. La variedad indiferenciada contiene células con importante pleomorfismo, con núcleos bizarros y una falta de cohesión característica. Desde el punto de vista ultraestructural, sólo algunos tumores presentan retículo endoplásmico liso y crestas mitocondriales tubulares, lo que subraya la dificultad de diferenciar este tipo tumoral de otros de su área anatómica, como el hipernefroma de células claras.
Desde el punto de vista diagnóstico, la potencial malignidad se ha basado en el análisis multifactorial de las características histológicas de tumores con evolución conocida, especialmente el peso, presencia de zonas extensas de necrosis, bandas de fibrosis, invasión capsular, invasión vascular, arquitectura del tumor (organizado o difuso), proporción de células claras, pleomorfismo nuclear, actividad mitótica de 5 * 50 HPF y presencia de mitosis atípicas. La presencia de tres de estas características permite afirmar el potencial de malignidad3. Recientemente, Evans y Vassilopoulou4 han sugerido que la presencia de cuatro figuras de mitosis por cada 10 HPF en las zonas más activas, la presencia de un patrón sólido o trabecular, la existencia de un considerable número de células con citoplasma eosinófilo y la necrosis son características fundamentales para confirmar el diagnóstico de carcinoma adrenal.
Desde el punto de vista inmunohistoquímico puede haber pérdida de tinción para citoqueratina, aumento de expresión para vimentina y, en algunos casos, tinción focal para la lectina Ricinus communis. Busan et al5 han comunicado que la inmunorreactividad para el anticuerpo A103, sustancia codificada por el gen Melan-A (MART-1), prácticamente excluye cualquier carcinoma que no siendo un melanoma pueda ofrecer dificultades diagnósticas con los tumores corticoadrenales.
El carcinoma cortical en ocasiones puede originarse de "células adrenales en reposo". Éstas han sido descritas en diversas localizaciones y, en forma de tejido adrenocortical accesorio o aberrante, se han localizado en el ligamento ancho, testículo (especialmente en niños) y en el polo superior del riñón. Más raramente se han localizado en posición intracraneal, hígado y pulmón. De todos modos, el desarrollo de tumores en estas localizaciones es excepcional, especialmente en sus formas hiperfuncionales, si bien hay que tenerlos en cuenta cuando se originan en el polo superior del riñón.
Biología
La biología del carcinoma adrenal se centra fundamentalmente en el mecanismo de la alteración de la esteroidogénesis y en la patogenia de este infrecuente tumor.
La esteroidogénesis se caracteriza por diversos fallos enzimáticos adquiridos durante la transformación neoplásica, que hacen que estos tumores secreten distintos tipos de precursores, muchos de ellos con escaso poder hormonal que hace que los síndromes endocrinos aparezcan sólo cuando el tumor adquiere un determinado tamaño. En segundo lugar, es característica la falta de respuesta a ACTH, y en tercer lugar lo que se ha querido denominar mecanismo de esteroidogénesis constitutiva.
Los carcinomas adrenales presentan a menudo un fallo enzimático en los sistemas de la 11-beta-hidroxilasa, pudiendo presentar también defectos de la 17-alfa-hidroxilasa, 21-hidroxilasa, 3-beta-hidroxiesteroide-deshidrogenasa y delta5-delta4-isomerasa. Por ello, aunque se acepte que la mayor parte son tumores no funcionantes, la realidad es que secretan distintos tipos de precursores que es necesario buscar. Así, se ha descrito la secreción de metabolitos de pregnenolona, tetrahidrodeoxicortisol, 11-desoxicortisol, pregnanediol, ocasionalmente testosterona no derivada del metabolismo periférico de la androstenediona, estrona, estradiol y desoxicorticosterona. Para caracterizar este tipo de secreción hormonal no vale la determinación de esteroides en sangre periférica o en orina, debiéndose de recurrir al estudio de gradientes hormonales por cateterización venosa6.
En segundo lugar, es raro observar casos de carcinomas adrenales que respondan al ACTH. Por ello se ha querido utilizar este defecto para diferenciar los tumores adrenales benignos de los malignos, lo que no es universalmente cierto, pues algunos carcinomas presentan discretos grados de respuesta. Además, se han comunicado casos de pacientes que presentando aún respuesta al ACTH in vivo, portaban tumores que no respondían al ACTH con la debida producción de AMPc o síntesis esteroidea, motivo por el que se deben interpretar con mucha cautela los estudios funcionales de estimulación con ACTH. Junto a los defectos enzimáticos es posible que se asocien defectos en los mecanismos de segundos mensajeros, pues se ha descrito que en cultivo hay tumores que demuestran una marcada síntesis esteroidea cuando son estimulados con análogos de AMPc que con ACTH. Es posible que el carcinoma adrenal presente también defectos en la expresión de receptores de ACTH o de fenómenos posreceptor, como pueden ser defectos en la función de sistemas proteincinasa o de translocación de colesterol.
El fenómeno de la esteroidogénesis constitutiva es, posiblemente, el hecho más característico de los tumores adrenales, lo que explica cómo un paciente puede tener valores suprimidos de ACTH mientras que, al mismo tiempo, se están produciendo cantidades suprafisiológicas de hormonas esteroideas. Al menos tres mecanismos genéricos pueden explicar este fenómeno. En primer lugar, la síntesis tumoral de esteroides podría simplemente reflejar el estado basal o no estimulado de una gran masa celular en el tumor, aunque no parece existir una proporcionalidad entre la síntesis de esteroides y la masa tumoral. Esta discrepancia podría ser corregida si se ajusta la síntesis hormonal a la masa celular activa, excluyendo el volumen necrótico tan frecuentemente observado, aunque parece que, debido a que los tumores producen menor cantidad de esteroides por unidad de masa, habría que aceptar que la esteroidogénesis tumoral constitutiva representa la esteroidogénesis basal, especialmente en tumores de gran tamaño. En segundo lugar, la esteroidogénesis podría ser estimulada por otros factores u hormonas que no tuvieran un papel predominante en condiciones fisiológicas normales, habiéndose descrito estimulación por PGE1 y agonistas betaadrenérgicos. Finalmente, la esterodogénesis constitutiva podría ocurrir debido a que procesos intracelulares distales fueran activados a través de mecanismos dependientes de activación por análogos de la vía AMPc-proteincinasa, mutaciones en esta vía, déficit en la acción de la fosfodiesterasa o por otros caminos alternativos (activación de la vía calcio-fosfatidilinositol), originando un proceso de activación tónica en ausencia de estimulación de hormonas tróficas6.
Desde el punto de vista patogénico, los esfuerzos se centran en el conocimiento de las alteraciones genéticas y sus posibles implicaciones3. Aunque los estudios son escasos, se ha demostrado una pérdida de alelos en los cromosomas 11p, 13q y 17p de carcinomas no presentes en casos de adenomas o hiperplasias, presentando generalmente un origen monoclonal7. Los tumores adrenales son más frecuentes en el síndrome de Gardner8, en el de Li-Fraumeni9 y en el de Beckwith-Wiedemann, donde también se ha demostrado una pérdida de heterozigosidad en el cromosoma 11p. Junto con la pérdida alélica de la región 11p13-15, que puede presentarse en el 78% de los cánceres corticoadrenales, se ha asociado la sobrexpresión del gen de la IGFII en el 80% de los tumores7,10 y alteraciones postranslacionales en la regulación de la proteína captadora del IGF (IGFBP-2) con aumentos no dependientes en la expresión de su ARNm11. Se han identificado mutaciones en la codificación genética de subunidades de la proteína G-inhibidora Gi2, que parece desempeñar un papel en los mecanismos de crecimiento y diferenciación y en su gen productor gip2, así como pérdida de heterozigosidad en el gen del receptor de la ACTH, que también podría contribuir en la desdiferenciación de adenoma y carcinomas12. El 52% de los tumores muestran positividad inmunohistoquímica para la p5313, con pérdida alélica para el gen que la sintetiza (17p) en el 61% de los casos y para el del retinoblastoma (RB)14 (80%), neuroblastoma (1p) (22%), von Hippel Lindau (3p) (22%) y gen p16 (9p) (26%)15.
Se ha demostrado la presencia inmunohistoquímica de receptores para el EGF en 63 de 64 carcinomas adrenales, lo que sugiere la posibilidad que el EGF o el TGF-alfa, que también es un ligando del receptor de EGF, puedan tener algún papel en la tumorogénesis adrenal junto al IGF-I e insulina, que parecen ser candidatos adicionales de una acción de regulación autocrina. Recientemente, Opocher et al16 han demostrado que la angiotensina II, que tiene efectos mitogénicos adrenales a través de la unión a los receptores de tipo I, expresa sus receptores en los adenomas secretores de aldosterona y en los de cortisol, pero no lo hace ni en el feocromocitoma ni en los carcinomas adrenales, por lo que la reducción de la expresión de estos receptores podría ser considerada como una prueba de desdiferenciación o malignidad de los tumores adrenales.
Clínica
La clínica de los carcinomas adrenales debe ser considerada en tres categorías: síntomas debidos a la propia masa adrenal, síntomas debidos a la diseminación locorregional y/o a distancia y síntomas endocrinos de hiperfunción17 (tabla 1).

Síntomas debidos a la masa adrenal
El tumor se palpa como una masa abdominal en el 30-40% de los pacientes a consecuencia de su lento crecimiento. Dependiendo del tamaño y localización puede producir síntomas gastrointestinales como saciedad precoz, sensación de distensión, náuseas y vómitos. Astenia, fatiga, pérdida de peso y febrícula son síntomas frecuentes, al igual que el dolor abdominal, ya por compresión de nervios adyacentes o por la reacción peritumoral secundaria a los episodios de necrosis y hemorragia. Ésta suele tener un inicio brusco, con dolor irradiado o no al flanco, frecuentemente acompañado de alteraciones neurológicas, y complicado por el desarrollo de íleo, hipotensión y shock o insuficiencia suprarrenal aguda si la glándula contralateral se encuentra suprimida por ser un carcinoma secretor de cortisol. La tendencia a invadir las estructuras vasculares es la causa de que se originen síntomas atípicos secundarios a trombosis de cava inferior (edemas periféricos), vena renal (hematuria y proteinuria) o tronco portal (hipertensión portal con hepatosplenomegalia). Además, la invasión venosa es la responsable de cuadros de embolismo con infarto pulmonar. Por todo ello se ha mantenido el aforismo de que los carcinomas suprarrenales pueden simular todos los síntomas del carcinoma renal, por lo que, si se tiene en cuenta que dos tercios de los pacientes presentan alteraciones en las pielografías intravenosas, no es difícil suponer que en muchas ocasiones este tumor sea confundido con el carcinoma renal.
Síntomas debidos a la invasión local o diseminación a distancia
Didolkar et al llamaron la atención de que un tercio de los pacientes eran atendidos a consecuencia de signos y síntomas relacionados con la aparición de metástasis. Nader et al comunicaron que de 77 pacientes, sólo el 3,9% mostraron una enfermedad localizada, mientras que el 72% desarrollaron metástasis que fueron clínicamente evidentes en el momento del diagnóstico en la mitad.
Localmente, el carcinoma adrenal invade los ganglios periaórticos, haciendo casi imposible la resección curativa. En el lado izquierdo es frecuente la invasión del diafragma y del páncreas, en el lado derecho la invasión del hígado, y en ambos lados la del riñón y la vena cava. Además, hay que tener en cuenta que, junto a la invasión del espacio retroperitoneal, algunos pacientes con tumor unilateral pueden presentar afectación sincrónica o metacrónica de la otra adrenal.
En términos de diseminación a distancia, el carcinoma suprarrenal metastatiza en el pulmón (60%), hígado (50%), linfáticos (48%), hueso (24%) y pleura y corazón (1%), siendo muy poco frecuente la metastatización a riñón (5%) y cerebro (4%). En la interpretación de los posibles focos metastásicos hay que tener cierta prudencia, pues estos pacientes muestran una tendencia a desarrollar segundos primarios, especialmente de mama, linfomas, tiroides, útero, colon y piel. Por ello, ante la presencia de un posible tumor primario oculto hay que pensar, entre otras posibilidades, en el carcinoma adrenal, descartándolo mediante TAC o RM.
Síntomas debidos a hipersecreción hormonal (tabla 2)
Aunque el número de tumores no funcionantes es superior en una relación de 4 a 1 no hay que olvidar las formas hiperfuncionales de presentación. Las formas funcionantes son más frecuentes en la mujer, casi un 75%, especialmente en la forma de presentación con hiperandrogenismo y no se correlaciona ni con la edad del paciente, tamaño del tumor ni presencia de metástasis.
Las endocrinopatías más frecuentes son la virilización en mujeres, el hipercortisolismo en ambos sexos, la feminización en el varón y la pubertad precoz. Menos frecuentemente pueden aparecer síntomas debidos a hipersecreción de mineralocorticoides, insulina, eritropoyetina, vasopresina, ACTH y Gh18. De todos modos, el carcinoma adrenal es el único tumor de esta glándula que puede originar, en vez de cuadros puros, cuadros mixtos de hipersecreción hormonal.
La producción ineficiente debida a fallos en las vías enzimáticas origina la secreción preponderante de esteroides biológicamente poco potentes, muchos de los cuales sólo pueden ser caracterizados mediante técnicas cromatográficas19 o análisis específicos. Bioquímicamente se traduce por un incremento en la secreción de 11-desoxicortisol, DHEA, estrona y 11-desoxicorticosterona, y clínicamente por el hecho de que se necesitan grandes producciones hormonales para que se produzca una expresión clínica (hiperfunción bioquímica sin hiperfunción clínica); que exista una relación directa entre el tamaño tumoral y la expresión clínica; que la presencia de grandes cantidades de precursores circulantes simule el perfil hormonal del síndrome adrenogenital virilizante, si bien la no supresibilidad a dexametasona y los hallazgos de la TAC pueden permitir la sospecha del carcinoma, y, por último, que la expresión clínica sea dependiente del tipo, potencia y magnitud de los precursores sintetizados, así como de la edad y sexo del paciente20.
Los síndromes más frecuentes a tener en cuenta son el síndrome de Cushing, el síndrome hiperandrogénico, el síndrome feminizante y el síndrome mineralocorticoide.
El síndrome de Cushing es uno de los más frecuentemente encontrados, especialmente en la mujer. La hipersecreción de glucocorticoides es autónoma y asociada a la tríada ACTH suprimida, sin respuesta a la supresión con altas dosis de dexametasona y sin respuesta a la metopirona, simulando las características de los adenomas adrenales. En ambos procesos se origina cara de luna llena, morrillo de búfalo, plétora, equimosis, hipertensión e hipopotasemia con igual frecuencia, mientras que el adelgazamiento cutáneo, las estrías y la hipertrofia ventricular izquierda son más típicas en los pacientes con adenomas. Característicamente, los pacientes con carcinoma presentan más signos de exceso androgénico como el aumento de grosor del pelo, la calvicie temporal, el acné y los diversos grados de franca virilización, a lo que se une la menor incidencia de miopatía secundaria a la neutralización del efecto catabólico de los glucocorticoides por el anabólico de los andrógenos. Por todo ello se ha afirmado que la asociación hipercortisolismo-hiperandrogenismo es patognomónica del carcinoma adrenal. Hormonalmente, la diferencia más simple entre el síndrome de Cushing secundario a adenoma y el secundario a carcinoma es la marcada elevación de la eliminación de 17-cetos en el carcinoma, que suelen superar los 15 mg/24 h, incluso en ausencia de signos evidentes de virilización.
Ya se ha comentado que la hipersecreción de andrógenos es uno de los acontecimientos más frecuentes en el carcinoma suprarrenal, pudiendo expresarse de formas muy distintas. En las mujeres adultas puede aparecer desde un hirsutismo a una virilización completa con amenorrea, hipertrofia del clítoris, aparición de barba, entradas temporales y calvicie. En el niño prepúber la manifestación más típica es el desarrollo de pubertad precoz isosexual, en las niñas pubertad precoz con hirsutismo y masculinización, mientras que en el varón adulto la hipersecreción androgénica puede pasar desapercibida y manifestarse tan sólo por un aumento de peso secundario al efecto anabolizante. Los fenómenos androgénicos, que también suelen estar condicionados por el tamaño tumoral debido a la escasa potencia de los andrógenos producidos, suelen ser mediados por la secreción de DHEA-S, pues la secreción de testosterona es rara. En combinación con la secreción de andrógenos no es infrecuente observar síndromes mixtos, especialmente bajo la asociación a hipercortisolismo, pudiendo exteriorizarse como síndrome de Cushing con androgenización, síndrome predominante de androgenización, escasa o nula androgenización pero con elevación marcada de la eliminación de 17-cetos o, finalmente, hiperandrogenismo acoplado a eliminación de 11-desoxicortisol, grupo de pacientes que se asemejan a aquellos con déficit de 11-beta-hidroxilasa.
Desde que en 1948 Armstrong y Simpson describieron el "feminismo adrenal" se sabe que, aunque raros, los carcinomas adrenales pueden originar feminización. La expresión clínica depende, una vez más, de la edad de aparición, sexo del paciente y magnitud de la secreción de estrógenos. Cuando ocurre en niñas prepuberales se origina una pubertad precoz seudoisosexual, cuando ocurre en mujeres adultas el efecto hiperestrogénico puede quedar enmascarado, mientras que en el varón adulto es típica la aparición de ginecomastia, pérdida de la libido e hipoandrogenismo.
La hipersecreción mineralocorticoide es rara, no representando más del 1-2% de todos los hiperaldosteronismos. No existen diferencias entre la edad de aparición, sexo y forma de presentación entre los adenomas y los carcinomas, pudiendo ser diferenciados según los datos proporcionados por la TAC, la hipersecreción de otras hormonas, la anatomía patológica y la presencia de metástasis. Desde el punto de vista hormonal, los carcinomas secretores de aldosterona se caracterizan por presentar un patrón funcional semejante al del adenoma, aunque la aldosteronemia suele ser mayor y la hipocaliemia más profunda en el carcinoma, elevación de precursores como la DOCA y la 18-hidroxicorticosterona, no suele observarse ritmo circadiano ni respuesta a la infusión de ACTH y, al contrario que los adenomas, los carcinomas no muestran el clásico descenso de aldosterona por los cambios posturales. Aunque se han descrito casos con secreción pura de aldosterona lo más frecuente es que la secreción tumoral sea mixta, fundamentalmente de cortisol y andrógenos y relacionada con la progresión del tumor. Asimismo, es excepcional la secreción pura y aislada de DOCA. Aupetit-Faisant el al21 han comunicado que la secreción de mineralocorticoides puede ser un marcador de malignidad en tumores asintomáticos o no secretores, pues en estos casos existe una disfunción secretora de la vía de la aldosterona que puede ser reconocida estudiando la relación entre aldosterona y el sustrato de la 11-beta-hidroxilasa, la DOCA, y su derivado la 18-OH-DOCA.
Diagnóstico y estadificación
Ante la sospecha de un carcinoma adrenal, solamente confirmable mediante estudio histológico, la secuencia diagnóstica se encamina a demostrar el grado de función, a identificar el grado de invasión local y delinear la extensión anatómica y a descartar la presencia de metástasis. El diagnóstico basado en el peso y tamaño de la tumoración, considerando potencialmente malignos los superiores a 6 cm o 50 g, de peso puede ser erróneo22.
Estudio hormonal
El estudio hormonal dependerá del tipo de síndrome endocrino sospechado. Aunque varias series quirúrgicas han indicado la prevalencia de los tumores no funcionantes, la realidad es que cuando se profundiza en el análisis de los esteroides secretados el número de tumores " hormonalmente activos " aumenta. Para ello se hace necesario el análisis cromatográfico de los distintos esteroides, lo que desde el punto de vista práctico es poco relevante pues al tratarse de precursores con escaso potencial biológico, no van a modificarse los planteamientos terapéuticos iniciales. Sin embargo, es aconsejable disponer del estudio cromatográfico preoperatorio, pues el perfil de esteroides puede ser un marcador evolutivo de la aparición de metástasis antes de su diagnóstico clínico23.
El diagnóstico de hipercortisolismo no difiere en nada del que se realiza en el síndrome de Cushing hipofisodependiente o en el originado por adenoma adrenal. Los únicos rasgos diferenciales entre el adenoma y el carcinoma suprarrenal son la elevación característica de los 17-cetosteroides en orina, la elevación de DHEA-S, el aumento de precursores circulantes y las alteraciones en los estudios de imagen.
La hipersecreción androgénica es puesta de manifiesto por la elevación de los 17-cetos urinarios o la de DHEA-S plasmática que, específicamente, puede ser el mejor marcador de carcinoma, en especial si sus concentraciones plasmáticas permanecen elevadas y no suprimidas a valores normales tras la supresión con 2 mg de dexametasona/día durante 5 días24. Las concentraciones plasmática de testosterona, total o libre, suelen ser poco reveladoras, y su aumento ocasional suele ser más frecuente en mujeres con tumores virilizantes.
La valoración de la secreción estrogénica sólo es necesaria cuando se demuestren en varones rasgos típicos de feminización o una pubertad precoz isosexual en niñas. Independientemente de la elevación de estradiol, estrona y/o estriol, es la regla observar un aumento de 17-cetos urinarios.
El estudio de la hipersecreción de aldosterona no difiere en nada del realizado en otros tipos de hiperaldosteronismo primario, si bien existen algunas diferencias con los adenomas que pueden hacer sospechar el diagnóstico. En general, los pacientes con carcinoma presentan cifras más elevadas de aldosterona plasmática, pierden el ritmo circadiano y postural de aldosterona y suelen presentar cifra más elevadas de DOCA y 18-hidroxicorticosterona. Por último, nuevamente la imagen suele ser resolutiva.
La determinación de precursores esteroideos tiene, quizás, más importancia académica que práctica. Puesto que el defecto enzimático más frecuente es el fallo de la 11-beta-hidroxilasa, se produce acumulación de 11-desoxicortisol que, tras ser convertido a 11-tetrahidro-11-desoxicortisol, es medido en la orina como 17-hidroxicorticode. Ésta es la razón por la que algunos pacientes con carcinoma adrenal pueden presentar un aumento en las concentraciones urinarias de 17-OH sin evidencia de síndrome de Cushing. Por tanto, la determinación de cortisol libre urinario o el fraccionamiento de los 17-OH pueden delimitar la naturaleza de la elevación de los 17-OH. La alteración de la 3-beta-hidroxiesteroide-deshidrogenasa puede originar una elevación en las concentraciones de pregnantriol, el metabolito urinario de la pregnenolona, razón por la que los 17-cetos pueden ser normales mientras que los esteroides 17-cetogénicos se encuentran elevados. De todos modos, no está claro si los tumores con aumento en la producción de metabolitos de pregnenolona deben ser denominados "tumores funcionantes".
Estudio radiológico
La primera técnica de imagen a utilizar en el paciente en quien se sospecha un carcinoma adrenal es la TAC o RM, seguida de la gammagrafía con I-colesterol que aporta una información adicional sobre la naturaleza funcional25. Otras técnicas invasivas como la arteriografía o la cavografía, pueden ser necesarias en ciertas ocasiones para definir algunos aspectos de utilidad para el tratamiento quirúrgico.
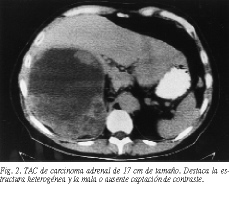
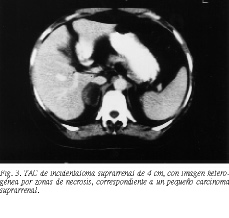
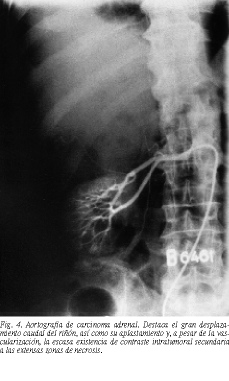
La TAC permite detectar la mayor parte de los carcinomas adrenales, siendo, cuando se valora el tamaño, la técnica más útil para diferenciarlo de los adenomas (fig. 2). En general, los carcinomas suelen ser mayores de 6 cm, especialmente en pacientes con signos de hipersecreción hormonal y/o demostración de invasión locorregional, presentan áreas de necrosis y/o calcificación aunque un pequeño porcentaje, alrededor del 6%, pueden ser menores (fig. 3). En pacientes con tumores no funcionantes, una masa adrenal superior a 6 cm es muy sospechosa de carcinoma, aunque no totalmente concluyente, debiéndose tener en consideración otros diagnósticos como quistes, los raros "adenomas grandes", lipomas y mielolipomas. En el caso de sospecha de un carcinoma no funcionante menor de 6 cm, la TAC por sí sola no permite asegurar el diagnóstico, por lo que en estos casos puede recurrirse a la RM y a la PAAF. La RM se caracteriza porque la intensidad de la señal en T2 respecto al hígado es mayor en el carcinoma, debiendo ser realizada siempre que se sospeche afectación de la cava26. Por último, la tomografía de emisión de positrones (PET) con desoxiglucosa o fludesoxiglucosa-F-18, que ha demostrado ser un magnífico método para diagnosticar las masas adrenales en pacientes con carcinomas conocidos27, deberá ser comparada con las dos técnicas clásicas, por lo que todo paciente con sospecha clínica de carcinoma adrenal debería ser estudiado con esta técnica. Otras técnicas como la arteriografía, muy útil en otros tiempos, prácticamente no tiene lugar en el armamentario diagnóstico actual (fig. 4).



La PAAF, realizada bajo control de imagen, es frecuente que no pueda asegurar el diagnóstico debido a las limitaciones de interpretación, a menos que los patrones histológicos sean muy típicos de benignidad o malignidad; por ello, no es una técnica ampliamente utilizada en el diagnóstico del carcinoma adrenal. De todos modos, Sharma at al28 han descrito un patrón citológico que puede facilitar el diagnóstico de carcinoma adrenal, especialmente cuando se pretende diferenciarlo del carcinoma renal. La presencia de una arquitectura endocrinoide con marcada anisonucleosis focal, la presencia de fragmentos de crecimiento alto y aplastado, los núcleos excéntricos y la ausencia de vacuolización citoplasmática son datos muy sugestivos de carcinoma adrenal. El carcinoma renal, por el contrario, suele presentar un patrón acinar, citoplasmas bien definidos y vacuolados, y si está presente el pleomorfismo es gradual y uniforme. Junto a ello, la presencia de mitosis, sólo observable en los tumores malignos, y la tinción positiva para la 3-beta-hidroxiesteroide-deshidrogenasa que permite identificar células adrenales parenquimatosas, puede ser de gran ayuda en el diagnóstico citológico29.
La gammagrafía con I-colesterol, incluso en casos de carcinomas "funcionales", suele demostrar un patrón de ausencia de captación secundario a la capacidad significativamente disminuida de captación del isótopo en el carcinoma respecto a la alta del adenoma. Del mismo modo, las metástasis tampoco suelen captar isótopo, si bien en algunos casos las metástasis funcionantes pueden transformarse en captadoras tras la extirpación del tumor primario. En ocasiones puede producirse una visualización simultánea del tumor primario y de sus metástasis. Recientemente se ha comunicado la localización inmunoisotópica con el fragmento Fab'2 del anticuerpo anticarcinoma adrenal Ac530.
Desde el punto de vista del diagnóstico diferencial, merece la pena recordar que el carcinoma suprarrenal debe tenerse presente ante cualquier masa retroperitoneal, especialmente si afecta al espacio renal-suprarrenal, ante cualquier incidentaloma adrenal no funcionante, en la evaluación de una enfermedad metastásica con tumor primario no conocido y ante cualquier gammapatía de significado incierto31. En el niño debe establecerse el diagnóstico entre el tumor de Wilms, neuroblastoma y nefroblastoma.
La estadificación, capital para realizar el planteamiento terapéutico, se ha basado en la propuesta en 1977 por el departamento de salud de los EE.UU. (SEER)32 y en la modificación introducida por Sullivan en 1978. Ambas consideran 4 estadios:
1. Enfermedad localizada: tumor que tras su extirpación está confinado a la adrenal no pudiéndose poner de manifiesto la existencia de metástasis. Menos del 5% de los carcinomas adrenales se encuentran en esta categoría.
2. Enfermedad regional: tumor con afectación del/los tejidos contiguos y de los órganos de vecindad, incluyendo los ganglios linfáticos, pero sin evidencia de metástasis a distancia. Entre el 20-30% de los tumores se incluyen en este grupo.
3. Enfermedad a distancia: tumores metastatizados en el momento del diagnóstico. En este grupo se incluyen el 40-60% de los tumores.
4. Enfermedad recurrente: aparición de recidiva local o diseminación a distancia tras la resección teóricamente curativa del tumor primario.
Sullivan33 considera la enfermedad local en dos grupos distintos de acuerdo con el tamaño tumoral, siendo la clasificación que se compara con la TNM:
Estadio I: (T1N0M0). Tumor menor de 5 cm sin ganglios, sin invasión local y sin metástasis.
Estadio II: (T2N0M0). Tumor mayor de 5 cm sin ganglios, sin invasión local y sin metástasis.
Estadio III: (T1 o T2N1M0 o T3N0M0). Cualquier tamaño, con ganglios o invasión local, sin metástasis.
Estadio IV: (cualquier T, cualquier NM1 o T3-T4N1). Cualquier tamaño, con ganglios o invasión local, con metástasis.
Recientemente, Icard et al34 han propuesto unificar los estadios I y II en uno solo definido como enfermedad local, con independencia del tamaño tumoral.
Pronóstico y evolución
En general el pronóstico es muy malo, con una mortalidad entre el 65-94% a los 5 años, una supervivencia media de 29 meses y una supervivencia media actuarial del 35% a 5 años34,35. Por estadios, la supervivencia actuarial a 5 años puede cifrarse en un 54%-46% para los estadios I-II, en un 43-21% para el III y en un 4-6% para el estadio IV36, aunque algunos autores, como Chapuis et al37, han comunicado supervivencias actuariales mayores en una serie en la que el porcentaje de resecciones curativas fue del 66% (del 78% para el estadio I, del 62% para el II y del 32% para los casos reoperados por recidiva local). La supervivencia no parece relacionarse con el sexo o la edad, aunque algunos autores indican un mejor pronóstico por debajo de los 40 años, o actividad hormonal, aunque parece que los tumores feminizantes y los raros productores de aldosterona tienen mejor pronóstico que los secretores de andrógenos y cortisol (52 y 52,5 meses para los primeros y 24 y 16,8 meses para los segundos35). El contenido de ADN, puesto que todos los tumores suelen ser aneuploides, tampoco tiene significado pronóstico38.
El pronóstico es peor en los tumores con índice mitótico mayor de 20 por 50 campos de alta magnificación (HPF) y, aunque casi todos los tumores presentan un patrón aneuploide con mayor tendencia a la metastatización39, no parece ser la regla, pues algunos no lo hacen y otros diploides diseminan a distancia. La expresión de p53 no se correlaciona con la significación pronóstica, no guardando relación ni con la supervivencia, ploidía o intervalo libre13. Recientemente, Tartour et al40 han comunicado que los tumores que presentan tinción positiva para el anticuerpo monoclonal D11 tienen una menor capacidad de metastatizar y una mayor supervivencia.
Tratamiento
Aunque el carcinoma adrenal es un tumor de alto grado de malignidad y una corta expectativa de vida, debe intentarse un tratamiento que al menos permita un tiempo aceptable de remisión, y en los casos de tumor funcionante, un mejor control de los síntomas41.
Cirugía
La cirugía, con visos curativos o paliativos, está indicada en los casos de tumores con enfermedad local o regional, mientras que en los pacientes con tumores diseminados puede ser discutible la indicación de cirugía paliativa de citorreducción, pudiendo ser nuevamente electiva en los casos de metástasis únicas aisladas en órganos asequibles y en los casos de algunas recidivas locales. En otras ocasiones, la cirugía de citorreducción está indicada para facilitar la acción de los fármacos adrenolíticos o de quimioterapia.
El planteamiento quirúrgico parte de una adecuada valoración del grado de extensión locorregional, para lo cual, independientemente de los datos aportados por la TAC o RM, puede ser necesaria la realización de estudios angioflebográficos que delimiten el aporte vascular, el drenaje venoso y la posible invasión de la cava (fig. 3). Estos estudios, en algunas ocasiones, pueden poner de manifiesto la presencia de afectación adrenal bilateral, lo que supone una indicación de adrenalectomía bilateral. La adrenalectomía laparoscópica está formalmente contraindicada ante cualquier tumor con sospecha de malignidad, pues se han descrito recidivas precoces fatales tras su realización42.
Es imprescindible una adecuada valoración de la función renal ante la eventual necesidad de realizar una nefrectomía en bloque con la tumoración adrenal, maniobra obligada en la mayoría de los casos que debe ir seguida de una linfoadenectomía regional de doble propósito, la adecuada resección oncológica y la posibilidad de realizar una correcta estadificación. Este proceder, aunque no parece que mejore el pronóstico final35, puede reducir, aunque no asegurar, la posible aparición de recidiva local, lo que en muchas ocasiones no es posible debido a la presencia de pequeños focos microscópicos de invasión o a la invasión de estructuras vitales. En estas ocasiones está indicada la extirpación de la mayor cantidad posible de masa tumoral (citorreducción), siempre que el riesgo quirúrgico no sea extremadamente alto. En casos seleccionados con invasión de cava puede estar indicada la trombectomía o la resección de la cava infrarrenal43, si bien los resultados son tan desesperanzadores que no parece ser una maniobra rutinariamente aconsejable44.
Este planteamiento quirúrgico, agresivo e iterativo34,45, puede estar indicado en casos seleccionados con recidiva local o metástasis única, especialmente si el intervalo libre es largo, pues mejora la supervivencia y permite obtener una mejoría de los síntomas en casos de tumores funcionales y una mayor efectividad de los fármacos adenolíticos46.
Tratamiento adrenolítico
El tratamiento postoperatorio con algunos fármacos de acción adrenolítica puede mejorar y prolongar la supervivencia cuando se instaura inmediatamente después de la cirugía, aunque no se sabe si este tratamiento puede prevenir la aparición de las recidivas locales.
El fármaco de elección es el o-p-DDD (Mitotane o Lisodren), utilizado para el tratamiento de la enfermedad metastásica, para mejorar los síntomas debidos al hipercortisolismo pues, al bloquear la 11-beta-hidroxilasa, reduce drásticamente la síntesis de cortisol, para intentar reducir la posible aparición de recidivas locales y para tratar, como única alternativa posible, la enfermedad con extensa diseminación a distancia1,47,48.
Desde la primera comunicación realizada por Bergenstal et al en 1959, el o-p-DDD ha sido ampliamente utilizado en el tratamiento de la enfermedad metastásica. Más recientemente, Hutter y Kayhoe en 1966, y Lubitz et al en 1973, han confirmado los primeros resultados comunicando remisiones mensurables del tumor en el 34 y 61% de los pacientes, respectivamente. De todos modos, el grado de respuesta clínica no supera el 25%48, aunque, de acuerdo con las experiencias contradictorias publicadas, los resultados pueden depender tanto del grado de variabilidad individual inherente a las propias metástasis o a la dosis utilizada. El que el tumor sea o no funcionante no influye en el grado de respuesta al mitotane, pudiendo estar más relacionada con su depósito en la grasa, hígado o cerebro, pues puede ser detectada en la sangre incluso meses después de haber cesado su administración. Éste es el motivo por el que la administración conjunta de glucocorticoides deba ser continuada después de la supresión del fármaco.
Puesto que no se conoce la concentración plasmática óptima, existe cierto grado de controversia respecto a las dosis que deben ser empleadas, si bien parece que las concentraciones plasmáticas efectivas deben ser superiores a 14 µ g/ml49,50 no sobrepasando los 20 µ g/ml a partir de los cuales ya se observa toxicidad neuromuscular. En la actualidad se recomiendan dosis más bajas que las inicialmente comunicadas, empezando con 1-2 g/día hasta llegar a 5-6 g/día frente a las iniciales de 8-10 g, lo que reduce la posible toxicidad del fármaco. Ésta, que es una de las mayores causas de su limitación terapéutica, se refleja en forma de náuseas (88%), vómitos (23%), diarrea (38%), debilidad, dermatitis (23%), ginecomastia (50%), artralgias (19%), leucopenia (7%), hipercolesterinemia, hepatotoxicidad, síntomas neurotóxicos (pérdida de memoria y capacidad de concentración (50%), somnolencia y vértigos) y prolongación del tiempo de hemorragia por un efecto semejante al de la aspirina.
El o-p-DDD también puede ser utilizado para reducir la cortisolemia y mejorar los síntomas derivados de la sobreproducción hormonal. A dosis entre 0,5 y 3 g/día produce atrofia adrenal y una abolición de la síntesis de gluco y mineralocorticoides, además de modificar el metabolismo periférico de los glucocorticoides y andrógenos. Con frecuencia se hace necesario aumentar las dosis para obtener el mismo efecto, por lo que en estos casos puede asociarse o ser sustituida por la aminoglutetimida o, mejor, por el ketoconazol, ajustando las dosis de la combinación de fármacos de forma individualizada.
En caso de obtenerse una buena respuesta hormonal (descenso del cortisol plasmático en un 50%) se puede esperar regresión tumoral en un 42% de los casos, mientras que prácticamente no se conocen regresiones sin descenso de las concentraciones de esteroides. Por ello, puede admitirse que las modificaciones de la cortisolemia a partir de las 2 semanas son el mejor índice para seguir la respuesta terapéutica. La valoración de la evolución se realiza a las 4-6 semanas de la iniciación del tratamiento. Si la enfermedad progresa se debe suspender y si la progresión es incierta debe mantenerse otras 4-6 semanas. La respuesta, en caso de producirse, tiene una duración media de 6-10 meses, siendo infrecuente la inducción de una nueva remisión. Éstas suelen ser parciales, aunque algunos autores han comunicado remisiones completas de larga duración, especialmente en niños.
En el momento actual puede, pues, afirmarse que el o-p-DDD prolonga en un 20-25% de los casos la supervivencia de los pacientes con carcinoma adrenal51. Desde la comunicación de Schteingart et al52 se había aceptado que, puesto que el grado de respuesta depende del momento de iniciar el tratamiento, el fármaco debe ser instaurado como terapéutica adyuvante antes de que se produzca una evidencia clínica de metástasis, o antes de realizar cirugía de una recidiva local si el paciente no estaba tratado previamente. Aunque Didolkar et al53 comunicaron la obtención de mejores resultados en mujeres, pacientes con tumores localizados, pacientes en los que se ha podido realizar una extirpación con visos de radicalidad curativa y en aquellos en los que el intervalo libre fue superior a 12 meses, en el momento actual el mayor grado de respuesta se ha obtenido en los casos de enfermedad metastásica34,54, no habiéndose comunicado resultados evidentes en los casos en los que pudo realizarse una cirugía radical50. Éste es el motivo, junto a la no existencia de estudios controlados frente a la publicación de casos puntuales55, de que el concepto de tratamiento instaurado precozmente tras la cirugía, a dosis bajas (1,5-2 g) y durante largo plazo puede ser muy discutible.
Quimioterapia
El carcinoma cortical es muy poco sensible a los fármacos de quimioterapia, y se ha afirmado que es un tumor intrínsecamente resistente como consecuencia de que la mayoría de ellos expresan el gen MDR56. No existen ensayos controlados que hayan comprobado el efecto que el mitotene ejerce sobre la disminución de expresión del gen MDR, aumentando la sensibilidad a los fármacos de quimioterapia debido al incremento de acumulación intratumoral. Tampoco existen ensayos sobre el efecto terapéutico del bloqueo de receptores de glucorticoides o progesterona, aunque se ha propuesto el tratamiento con RU486 después del fallo con mito tane41.
Los principales protocolos utilizados con los que se han obtenido algunas remisiones totales de un año o más de evolución combinan varios fármacos. Se han basado en la utilización de doxorubicina y agentes alquilantes, siendo al parecer más prometedoras las combinaciones de cisplatino y etopósido, de 5-fluororuracilo, doxorubicina y cisplatino o de mitotane asociado a etopósido, doxorubicina y cisplatino41,57. Se ensayan nuevos agentes como es la suramina, que inhibe la unión del factor de crecimiento epidérmico, factor de crecimiento derivado de las plaquetas y el factor transformador de crecimiento tipo beta58, así como el Taxol8.
Radioterapia
La radioterapia tiene sólo un papel paliativo, especialmente en las metástasis óseas dolorosas41, aunque de acuerdo con la reciente experiencia de Crucitti et al35 habría que reconsiderar el papel de la radioterapia intra y postoperatoria.
Carcinomas corticales en niños
La presencia de un tumor cortical en un paciente menor de 16 años o en un niño es muy probablemente un carcinoma59-62, cuya incidencia se ha cifrado en un 66%. Como en el adulto, suelen ser tumores mayores de 4 cm, con una preponderancia masculina de 2/1 y una edad media de presentación de unos 5 años. La forma más frecuente de presentación es el hirsutismo (50%) y la virilización (76%), síndrome de Cushing (30%) y feminización (10%), tumoración adrenal no funcionante (8%) o muy raramente asociado con un síndrome de pérdida salina por hiperplasia adrenal congénita. No son infrecuentes las asociaciones con malformaciones de otros órganos, como la hemihipertrofia, síndrome de Beckwith-Wiedemann, malformaciones vasculares, urológicas o tumores cerebrales. El 57% tienen tumor palpable, el 80% enfermedad local, el 7% enfermedad locorregional y el 13% metástasis a distancia en el momento del diagnóstico. El estudio bioquímico no se diferencia del observado en el adulto y las normas terapéuticas son las mismas, si bien no existen pruebas de la eficacia de la quimioterapia. Como en el adulto, la supervivencia media es del 50%, aproximadamente, a los 5 años.
Carcinoma de la médula suprarrenal
Clásicamente se ha reconocido que su incidencia es del 10% de todos los feocromocitomas, pero datos muy recientes recopilados por Norton58 indican que, con un seguimiento prolongado, estudio meticuloso de las recidivas y metástasis, esta cifra puede aumentar hasta un 30 o un 50%, habiéndose descrito la aparición de metástasis a los 29 años de la intervención primitiva. De hecho, algunos autores como Beierwaltes, Sisson y Shapiro recomiendan la realización sistemática de gammagrafía con MIBG después de la extirpación de cualquier feocromocitoma esporádico. Son más frecuentes en los de localización ectópica, sobre todo en los de vejiga.
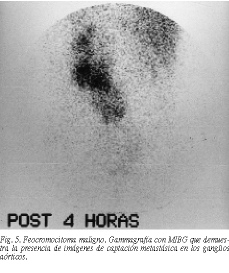
Desde el punto de vista clínico y diagnóstico, no se diferencian en nada de los feocromocitomas esporádicos, aunque en estos casos la gammagrafía con MIBG puede poner en evidencia la presencia preoperatoria de metástasis (fig. 5). La pre sencia de concentraciones plasmáticas elevadas de enolasa neuronal específica puede ser un marcador predictivo de malignidad63. Los criterios de malignidad son los mismos que en el carcinoma cortical, aunque en algunas ocasiones son más difíciles de definir. La cirugía radical con linfoadenectomía es el único tratamiento que puede llevar a la remisión completa, incluso en los casos con afectación de la cava inferior pues, al contrario que en los carcinomas corticales, el pronóstico es mejor. La cirugía de citorreducción metastásica debe intentarse siempre que se pueda64.

Esta terapéutica agresiva se ha basado en el posible tratamiento de la enfermedad residual con altas dosis de MIBG, aunque los resultados obtenidos no han sido tan espectaculares como se había supuesto65. La quimioterapia con altas dosis de estreptozotocina sola o ciclofosfamida asociada a vincristina y darcarbacina ha originado respuestas parciales del 50% a los 18 meses a cambio de una alta neurotoxicidad, y con una predicción de respuesta que se correlaciona con los valores preoperatorios de noradrenalina plasmática.

