




Introducción
El carcinoma de paratiroides se presenta comúnmente en pacientes con hiperparatiroidismo primario. Es muy rara su descripción en pacientes con hiperparatiroidismo secundario o terciario1 y se ha descrito sólo en 17 casos en la literatura médica, en una revisión completa de los casos registrados; el caso actual es el número 182.
Caso clínico
Paciente varón de 53 años de edad referido para el tratamiento quirúrgico de un hiperparatiroidismo terciario en 1999. Presentó inicialmente hipertensión arterial grave y proteinuria con el fracaso renal 10 años antes (1989), por lo que fue programado para diálisis.
Recibió un trasplante renal en abril de 1990. A pesar del tratamiento con análogos de la vitamina D y a causa de la persistencia de su hiperparatiroidismo, debido a dolor óseo severo y prurito fue programado para paratiroidectomía. Se realizó una paratiroidectomía total con autotrasplante en el músculo esternocleidomastoideo derecho, técnica empleada ocasionalmente en nuestro hospital y actualmente desechada. El diagnóstico histopatológico de las paratiroides extirpadas y previo al trasplante fue hiperplasia difusa paratiroidea, no nodular. El 29 de octubre de 1990 ingresa por deterioro de la función renal, que se interpretó como crisis de rechazo agudo y se trató con ciclo de Solumoderin a dosis de 1 g durante 3 días consecutivos, pero no presentó respuesta; en el segundo ciclo de Solumoderin a dosis de 500 mg durante 3 días consecutivos se obtuvo una respuesta parcial. Se realizó una biopsia renal (el 9 de noviembre de 1990) informada como rechazo agudo grave. Se trató con un tercer ciclo de Solumoderin sin que presentara respuesta, por lo que se decidió tratar con OKT3 a dosis de 5 mg, durante 10 días consecutivos. La creatinina final de 2,3 a 3,1 mg/dl. En su evolución posterior presentó deterioro de la función renal durante 1991, con una creatinina de 6,5 mg/dl, valor en el que se mantiene con discretas oscilaciones hasta 1995, cuando se produce un deterioro lento y progresivo hasta alcanzar una creatinina de 11 mg/dl en julio de 1998, por lo que se reinició el tratamiento con hemodiálisis periódicas.
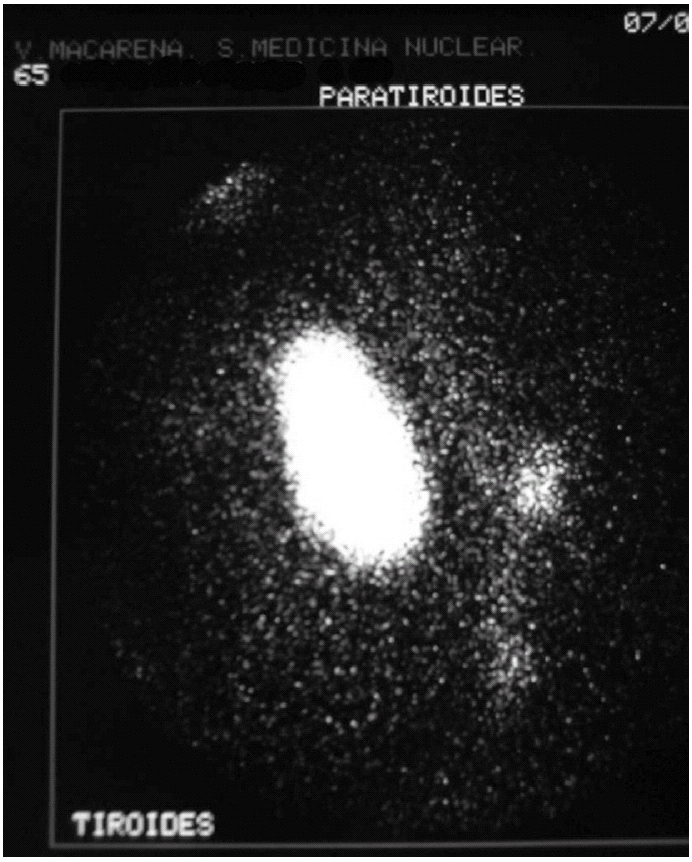
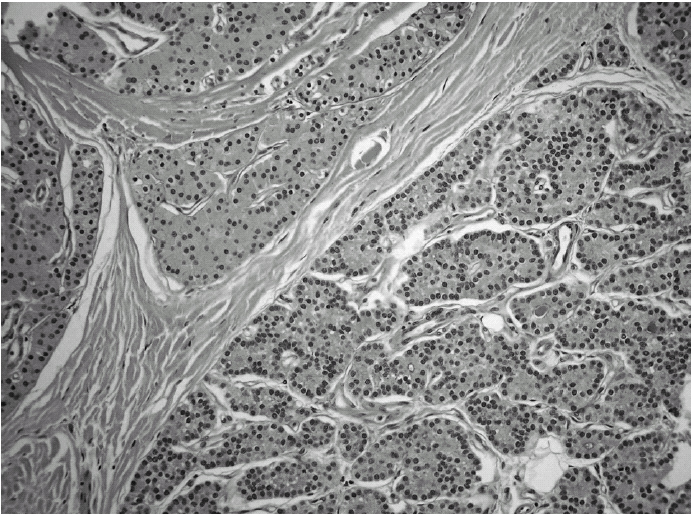
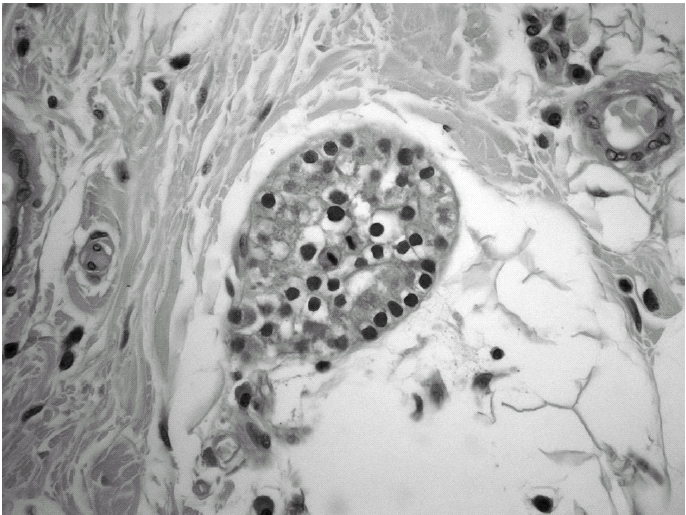
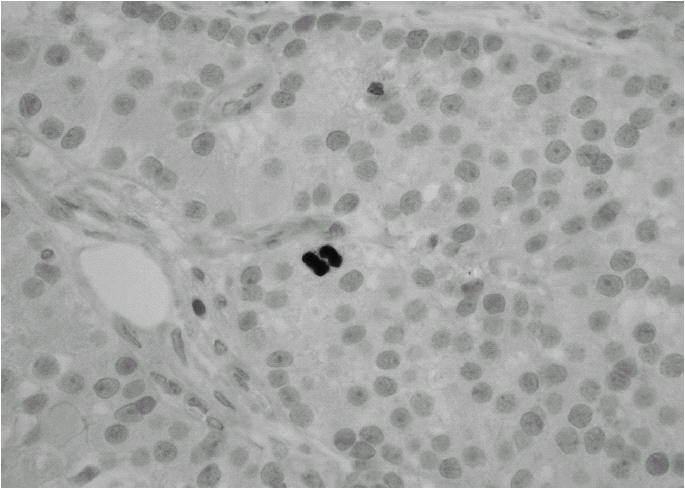
Durante el seguimiento mostró una elevación progresiva de PTH, que alcanzó valores de 886,5 pg/ml (normal < 60 pg/ml). El calcio sérico en ese momento era de 10,6 mg/dl (normal, 8,4 a 10,2 mg/dl). En la exploración clínica se apreciaba una tumoración cervical aparente en el área tiroidea derecha. No existían adenomegalias cervicales, supraclaviculares o axilares evidentes. La ultrasonografía cervical (fig. 1) reveló una masa hipervascular y extratiroidea (de 3,3 × 2,3 cm) y una imagen hipoecoica más profunda en la zona del hemitiroides izquierdo. La gammagrafía con tecnecio-99 sestaMIBI para examen cervical y mediastínico reveló un marcado aumento de captación del radiotrazador por la tumoración clínicamente evidente. No existía captación cervical. Se realizó una exéresis de la tumoración incluida en el músculo esternocleidomastoideo con resección parcial de éste y en el área del anterior trasplante y se complementó con una hemitiroidectomía del mismo lado por la existencia de adherencia firme de la tumoración a ésta. El examen histopatológico demostró macroscópicamente áreas de cambios degenerativos, hemorragia extensa y necrosis. Existía encapsulación fibrosa gruesa, invasión tumoral capsular, rabeculación y septos fibrosos, formación de rosetas epiteliales, pleomorfismo nuclear y mitosis visibles (figs. 2, 3 y 4). Un rastreo con tecnecio-99 sestaMIBI no demostró otros focos de captación. El paciente se encuentra con valores bioquímicos normales pasados 24 meses.
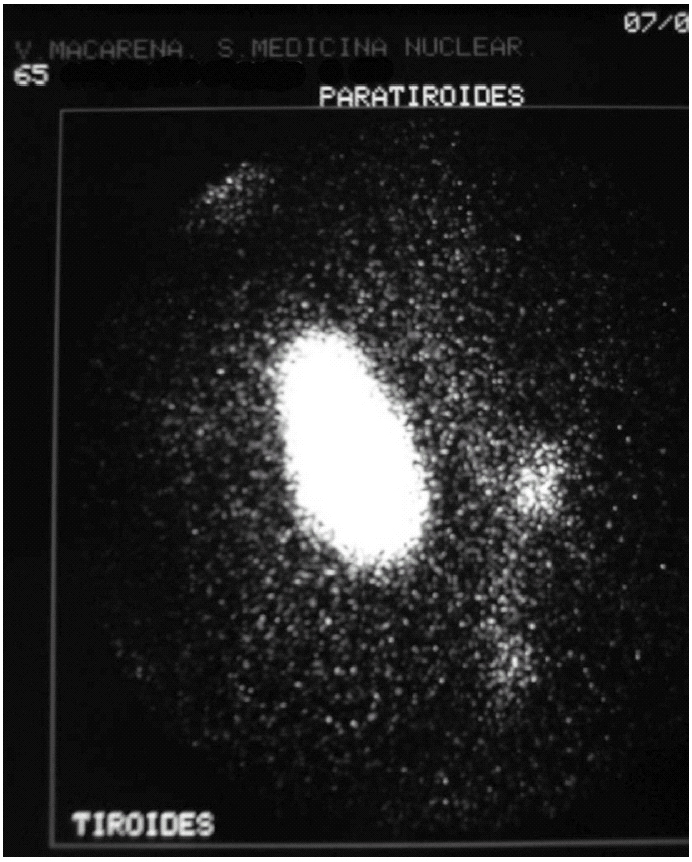
Fig. 1. Escáner con tecnecio-99 sestaMIBI. Captación masiva en la zona de la tumoración cervical y pequeña imagen adyacente.
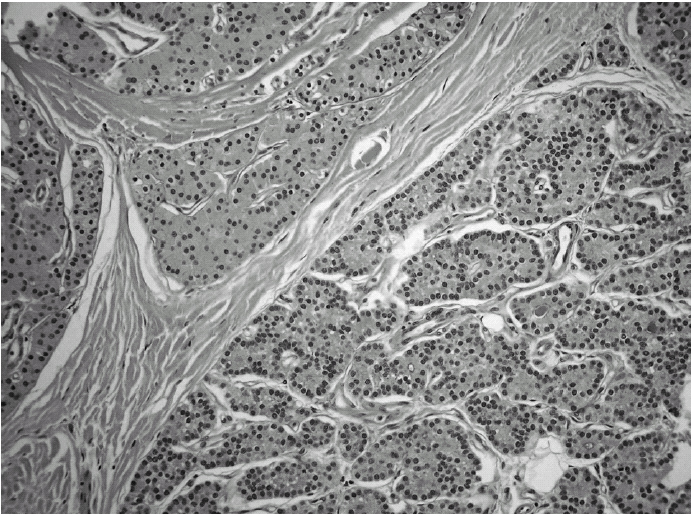
Fig. 2. Trabeculación fibrosa. (Hematoxilina-eosina, ×200).
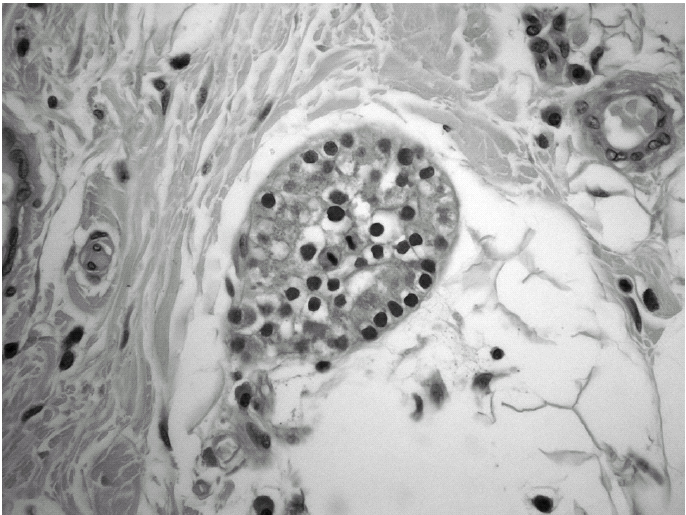
Fig. 3. Anomalías y pleomorfismo nuclear. (Hematoxilina-eosina, ×300).
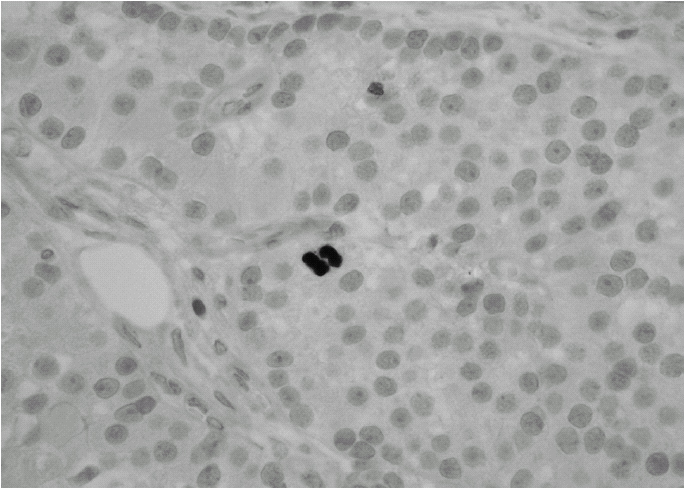
Fig. 4. Evidencia de mitosis activa. (Hematoxilina-eosina de Mayer, ×400.)
Discusión
Virtualmente todos casos de cáncer de paratiroides están asociados a situaciones de hiperparatiroidismo primario. La hiperplasia y proliferación celular paratiroidea habitualmente acompañan al fracaso renal crónico en el que la retención de fósforo va seguida de una disminución plasmática de calcio con secreción aumentada de paratohormona3. En sólo 18 pacientes se ha descrito el desarrollo de un carcinoma de paratiroides en situaciones de hiperparatiroidismo secundario o terciario en el enfermo renal crónico2. El primer caso se publicó en 1982. En el momento del diagnóstico, 13 de los 17 pacientes se presentaron con hiperparatiroidismo terciario. Se han descrito elevaciones de calcio en suero de hasta 24 mg/dl y valores de PTH de 98.000 pg/ml en alguno de estos pacientes. En nuestro paciente el valor preoperatorio de PTH no excedió de 900 pg/ml. Ningún indicio preoperatorio o postoperatorio era indicativo de encontrarnos ante un carcinoma de paratiroides. Dos de los 18 pacientes descritos mostraron dos glándulas con carcinoma y en los restantes solamente una glándula estaba afectada. La supervivencia general para el cáncer paratiroideo en enfermos no renales se ha estimado entre el 69 y el 85% en 5 años, y entre el 56 y el 70% a los 10 años4. La supervivencia a los 5 y los 10 años en carcinoma recurrente es del 50 y el 35%, respectivamente.
En los casos de enfermos renales en diálisis, la supervivencia registrada más prolongada es de 9,5 años5. Aún en la era de la terapia con análogos de vitamina D, un número significativo de pacientes requiere todavía paratiroidectomía para el tratamiento de las complicaciones evolutivas. En la mayoría de las glándulas de estos pacientes se encuentra una afectación hiperplásica, bien difusa, bien policlonal y ocasionamente monoclonal, sobre todo en pacientes de larga evolución y afectación grave6. Estas transformaciones pueden implicar mutaciones somáticas, tal como la pérdida alélica del gen WT1 supresor. La causa por la que estas glándulas pueden experimentar una transformación maligna es desconocida. Se han descrito otras anomalías genéticas tales como anomalías del gen supresor del retinoblastoma7.
Estos cambios no se describen en los adenomas de naturaleza benigna.
La existencia de un carcinoma de paratiroides es muy rara en pacientes de diálisis. Se ha sugerido que podría ser más común de lo esperable por el azar. Por otra parte, cerca de un 10% de todos los cánceres de paratiroides no son funcionantes y de ello se sigue un diagnóstico tardío por la tardanza en la manifestación de signos y síntomas8.
El diagnóstico diferencial con la proliferación excesiva del tejido trasplantado formas seudotumorales y la paratiromatosis deben considerarse en todos los casos antes de establecer el diagnóstico de carcinoma.
El tratamiento quirúrgico óptimo del cáncer paratiroideo implica una resección agresiva de la glándula como actitud inicial, lo que ofrece mejores posibilidades de curación. Existen varias características macrocópicas típicas que quizá pueden ayudar al cirujano a reconocerlo en el momento de la operación9. Éstas incluyen: el tamaño, un color blanquecino o gris del tumor y que sea firme y con adherencia al tejido circundante. Es muy importante no romper la cápsula del tumor. La aspiración con aguja fina y la simple biopsia incisional, así como la resección incompleta, pueden causar siembra local de células tumorales. La exéresis en bloque con tejido adyacente, cuando sea posible, es el procedimiento de elección. Si existe afectación linfática debe asociarse una linfadenectomía.
Patológicamente, el patrón oro para diagnosticar un carcinoma paratiroideo requiere la evidencia de la invasión del tumor en tejidos normales circundantes, la invasión vascular, metástasis de ganglios linfáticos, o metástasis a distancia que suelen ocurrir en el pulmón, en el hueso, en el hígado o en el cerebro. Hay varios patrones histológicos indicativos bien descritos: la encapsulación fibrosa gruesa, la invasión tumoral de la cápsula, la existencia de trabeculación y los septos fibrosos gruesos dentro del tumor, la formación de rosetas epiteliales en la arquitectura celular, el aumento de tamaño y pleomorfismo nucleares, y la presencia de imágenes mitóticas10. Varias de estas características se pueden observar en el tejido de nuestro paciente.
Si se demuestra la existencia de enfermedad metastásica, el tratamiento debe enfocarse a bajar el calcio y la PTH, pero la oportunidad de curación generalmente es limitada. La paliación más efectiva es la eliminación quirúrgica de cuanto tejido tumoral sea posible, si bien el empleo de biofosfonatos puede lograr una reducción temporal de los valores de calcio. La mortalidad por cáncer de paratiroides ocurre raramente debido a la metastatización tumoral en sí misma, sino que es generalmente el resultado de secreción excesiva de PTH, con producción de desajustes metabólicos, fundamentalmente la hipercalcemia y sus consecuencias. Los regímenes actuales de quimioterapia y radiación no han ofrecido resultados positivos. Debido al escaso número de casos descritos en el hiperpartiroidismo renal solamente podemos especular en cuanto a los factores de riesgo que puedan favorecer su desarrollo: la radiación externa previa, el estímulo continuado de la hipocalcemia o los factores genéticos pueden desempeñar un papel. El cambio maligno de un adenoma o hiperplasia a adenocarcinoma, secundario a la uremia, puede ser también una posibilidad. Hay una asociación muy conocida, pero infrecuen-te, entre el hiperparatiroidismo familiar, como la parte de neoplasia endocrina múltiple tipo 1, y el carcinoma de paratiroides.
Correspondencia:
Dr. M. Echenique-Elizondo.
Catedrático de Cirugía. Universidad del País Vasco.
Departamento de Cirugía. UD de Medicina.
P. Dr. Begiristain, 105.
20014 San Sebastián. España.
Correo electrónico: gepecelm@sc.ehu.es
Manuscrito recibido el 25-1-2005 y aceptado el 5-4-2005.

