




Los mecanismos epigenéticos fisiológicos que pueden modificar la estructura de la cromatina comprenden la metilación del ADN, las modificaciones de las histonas y las modificaciones en el RNA. Se ha llevado a cabo una revisión bibliográfica en PubMed acerca de la evidencia publicada sobre la relación existente entre la epigenética y el cáncer colorrectal. La literatura científica pone de manifiesto que en el origen de la enfermedad neoplásica las alteraciones epigenéticas pueden ser tan importantes como las modificaciones genéticas, contribuyendo ambas a la progresión y desarrollo de la enfermedad neoplásica.
The epigenetic and physiological mechanisms that alter the structure of chromatin include the methylation of DNA, changes in the histones, and changes in RNA. A literature review has been carried out using PubMed on the evidence published on the association between epigenetics and colorectal cancer. The scientific literature shows that epigenetic changes, such as genetic modifications may be very significant in the origin of neoplastic disease, contributing both to the development and progression of the disease.
El término «epigenética» fue acuñado por C.H. Waddington, para referirse al estudio de las interacciones entre los genes y el ambiente1. Paulatinamente a la epigenética se ha ido implicando una amplia variedad de procesos biológicos, definiéndose actualmente como el conjunto de cambios heredables de la expresión genética que ocurren de forma independiente a los cambios en la secuencia primaria de los nucleótidos del ADN2,3.
Aunque los cambios epigenéticos pueden ser reversibles, la mayoría de estos cambios heredados se mantienen estables a través de múltiples ciclos de la división celular, permitiendo a las células tener identidades diferentes mientras contienen la misma información genética. La herencia de los patrones de expresión genética es mediada por las modificaciones epigenéticas, por lo que sus alteraciones pueden inducir modificaciones de diversas vías de señalización y permitir el establecimiento de enfermedades como las neoplasias. Se ha postulado que estos cambios epigenéticos podrían ser el evento iniciador en algunos tumores2,3.
La estructura de la cromatina define el estado en el que la información genética está organizada en la célula2. Los mecanismos epigenéticos fisiológicos que pueden modificar la estructura de la cromatina se denominan epigenoma, y comprenden la metilación del ADN, las modificaciones de las histonas y las modificaciones en el ARN (fig. 1). La interacción entre estas modificaciones regula la vía por la que se manifiesta el genoma en los distintos tipos celulares, estadios de desarrollo y enfermedades. Por lo tanto, mientras que el código genético proporciona toda la información de los elementos celulares, el código epigenético controla la expresión de dicha información2,4.
Métodos
La revisión bibliográfica sobre la epigenética y el cáncer colorrectal se realizó en PubMed, iniciándose la búsqueda mediante el término Mesh «Epigenomics [Mesh] AND Colorectal Neoplasms [Mesh]». Con posterioridad, y debido al escaso número de artículos encontrados, se modificó la línea de búsqueda mediante los términos «Epigenetic AND «Colorectal Neoplasms»[Mesh]», comprobándose que todas las referencias obtenidas en la primera búsqueda estaban contempladas en los resultados posteriores. Ambas líneas de búsqueda se realizaron sin ningún tipo de limitación. La búsqueda reportó un total de 658 artículos, de los que se seleccionaron aquellas publicaciones que aportaban la información relevante para el desarrollo del presente trabajo.
Epigenética de la enfermedad neoplásicaLas células neoplásicas albergan tanto alteraciones epigenéticas como modificaciones genéticas que interactúan en todas las fases del desarrollo tumoral, permitiendo de forma conjunta el progreso de la enfermedad. El cáncer colorrectal ha servido como prototipo para el estudio de ambos cambios genéticos y epigenéticos, debido al amplio rango de lesiones patológicas y a que los cambios epigenéticos persisten a lo largo de toda su progresión2,3. La epigenética de la enfermedad neoplásica se caracteriza por la metilación del ADN, por la modificación de los patrones de las histonas y por la alteración del perfil de expresión de las enzimas que modifican la cromatina. Estas modificaciones epigenéticas van a repercutir sobre genes involucrados en el ciclo celular, en la reparación del ADN, en la apoptosis, en la angiogénesis, en la invasión y en la adhesión. De forma global, los cambios epigenéticos resultan en una disregulación de la expresión genética que permite el desarrollo y progresión de la enfermedad. Estas epimutaciones pueden promover la carcinogénesis mediante dos mecanismos: 1) silenciando los genes supresores de tumores, ya sea de forma independiente ya sea de forma conjunta, con mutaciones genéticas o deleciones; 2) mediante la activación de los oncogenes como consecuencia de la metilación aberrante del promotor, o por alteraciones genéticas adquiridas. Los genes supresores de tumores regulan de forma negativa la proliferación celular, por lo que las mutaciones a nivel de estos genes provoca una inactivación de los mismos que permite el crecimiento tumoral. Por el contrario, la mutación de los proto-oncogenes puede dar lugar a los oncogenes, permitiendo la proliferación celular por su capacidad carcinogenética2,5–7.
Diferentes estudios ponen de manifiesto que junto a la metilación del ADN y la modificación de las histonas, la remodelación de los nucleosomas desarrolla un papel esencial en el silenciamiento de los genes específicos de tumores. La metilación inducida del ADN para el silenciamiento de los genes supresores de tumores implica diferentes cambios en el posicionamiento de los nucleosomas, haciendo que estos se localicen en los sitios de inicio de transcripción2,7.
Vías celulares y epigenéticaEl cáncer es una enfermedad compleja que afecta a múltiples vías celulares tales como el ciclo celular, la reparación del ADN, la apoptosis, la respuesta hormonal, la invasión celular tumoral y las metástasis. La inestabilidad genética se caracteriza por la acumulación de mutaciones cromosómicas y cambios epigenéticos de los genes implicados en la regulación del crecimiento celular. Se ha evidenciado que los cambios epigenéticos que regulan dichas vías celulares conlleva la activación los oncogenes, la inactivación de los genes supresores de tumores, de los receptores hormonales, de los receptores de citoquinas, de la inactivación de genes de micro-ARN y de los receptores de los factores de crecimiento5,8.
Una de las características más importantes de las células neoplásicas es su capacidad para emigrar e invadir otros órganos a través de la vía sanguínea. Algunas de las proteínas que intervienen en este comportamiento tumoral son Adenomatous polyposis coli (APC), E-cadherina (CDH1) y H-cadherina (CDH13); característicamente los promotores de las mismas se encuentran hipermetilados. Junto a estas alteraciones epigenéticas, en las células tumorales existen también otras alteraciones como mutaciones en el oncogen K-ras, APC, TP53, BRAF, gen de la β-catenina, TP53, así como inestabilidad de los microsatélites que conlleva mutaciones en las vías de reparación del ADN dañado y pérdida de heterocigosidad4,9–11.
El gen APC es el más estudiado en el cáncer colorrectal, y mientras las mutaciones somáticas conllevan una alteración en la regulación de la expresión de la β-catenina (y por tanto la activación de la vía Wnt), la metilación aberrante del promotor del gen APC está asociada con la inestabilidad de los microsatélites a nivel de 5q, e implica un silenciamiento en la transcripción genética12,13.
Metilación del ácido desoxirribonucleicoLa metilación del ADN proporciona un mecanismo de silenciamiento de genes, bien por la prevención o bien por la promoción del reclutamiento de las proteínas reguladoras del ADN, que determina una función importante en la regulación de la expresión genética y en la arquitectura de la cromatina2,4.
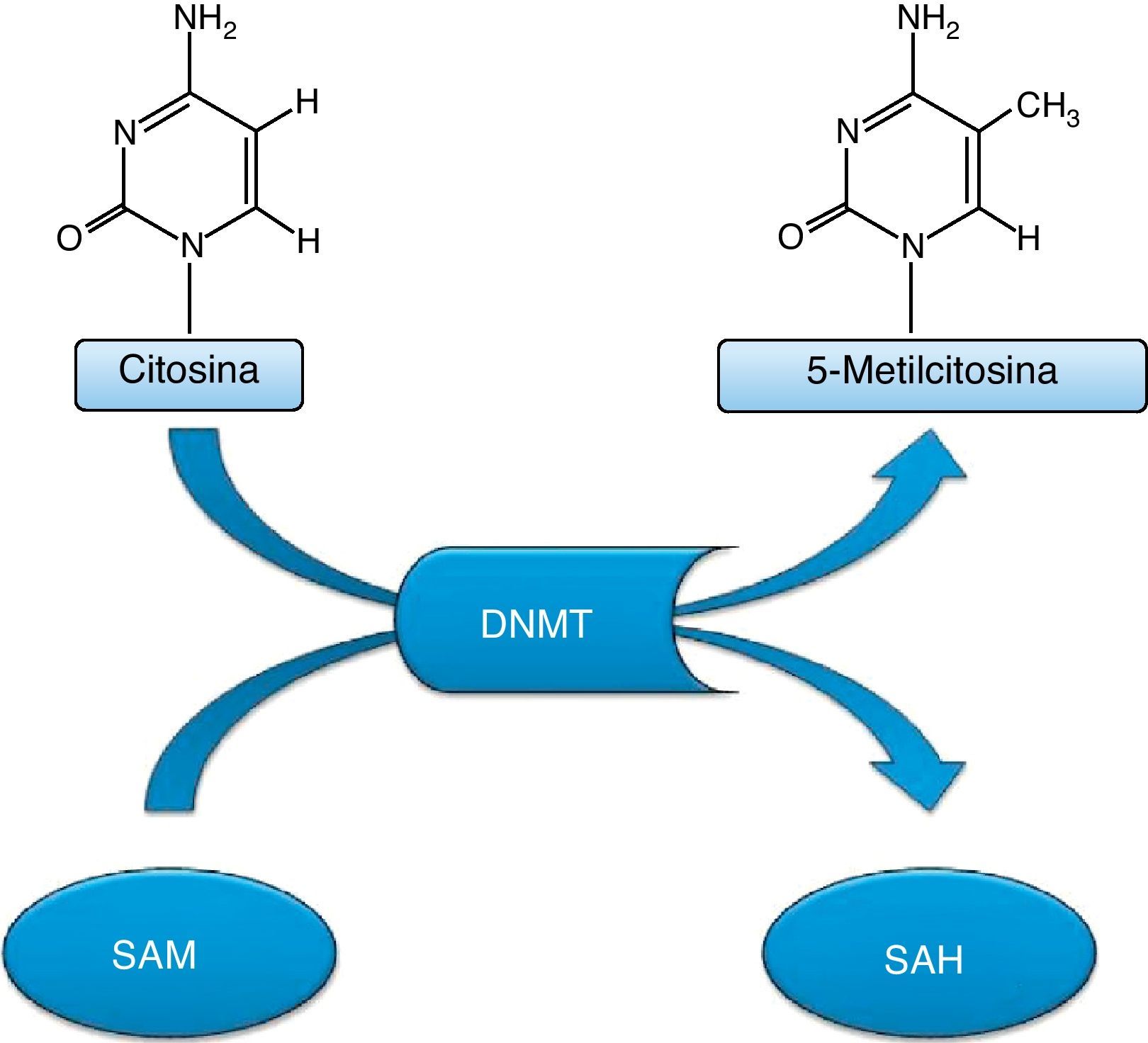
Esta metilación se lleva a cabo mediante la modificación covalente de los residuos de la citosina a nivel de los dinucleótidos 5′-CG-3′, también denominados dinucleótidos CpG (caracterizado por que a un nucleótido de citosina le sigue un nucleótido de guanina, enlazados por un fosfato); la distribución de estos nucleótidos no es uniforme en el genoma humano, pues mientras aproximadamente el 70% se localizan de forma dispersa, el 30% restante se localizan en regiones denominadas CpG islands o islotes CpG. El resultado de este proceso bioquímico es la formación de una base inestable, por lo que la metilación del ADN puede considerarse como un mutágeno endógeno (fig. 2)3,4,7,14.

El proceso de metilación del ADN se caracteriza por ser una reacción reversible que es catalizada por ADN metiltransferasas, de las que se diferencian tres tipos: DNMT1, DNMT3a y DNMT3b3. El epigenoma del cáncer está marcado por la hipometilación global del ADN y por la hipermetilación de los promotores genéticos a nivel de CpG islands14,15.
La característica reversibilidad de la metilación del ADN ha dado lugar al desarrollo de fármacos que inducen su desmetilación; como la 5-aza-2′-deoxicitidina que puede conducir a la reexpresión de genes silenciados. Estos agentes se incorporan al ADN de las células con alto índice mitótico, inhibiendo irreversiblemente la actividad de las ADN metiltransferasas y previniendo la hipermetilación de los islotes CpG16.
Metilación aberrante del ácido desoxirribonucleicoEl nivel de hipometilación global del genoma está relacionado con el grado de malignidad, por lo que podría servir como marcador biológico con valor pronóstico. La pérdida global de la metilación podría tener, al menos, tres consecuencias: la activación de la transcripción de oncogenes, la activación de retrotransposones latentes y la inestabilidad cromosómica. Asimismo se ha descrito el papel de la desmetilación en la reactivación de secuencias correspondientes a micro-ARN habitualmente silenciados17.
Además, la hipometilación permite la activación de genes que promueven el crecimiento, como puede ser el S-100 en el carcinoma colorrectal. Por lo tanto, la hipometilación del ADN permite la activación aberrante de genes y de regiones no codificantes, a través de una gran variedad de mecanismos que contribuyen al desarrollo de la enfermedad tumoral2,15.
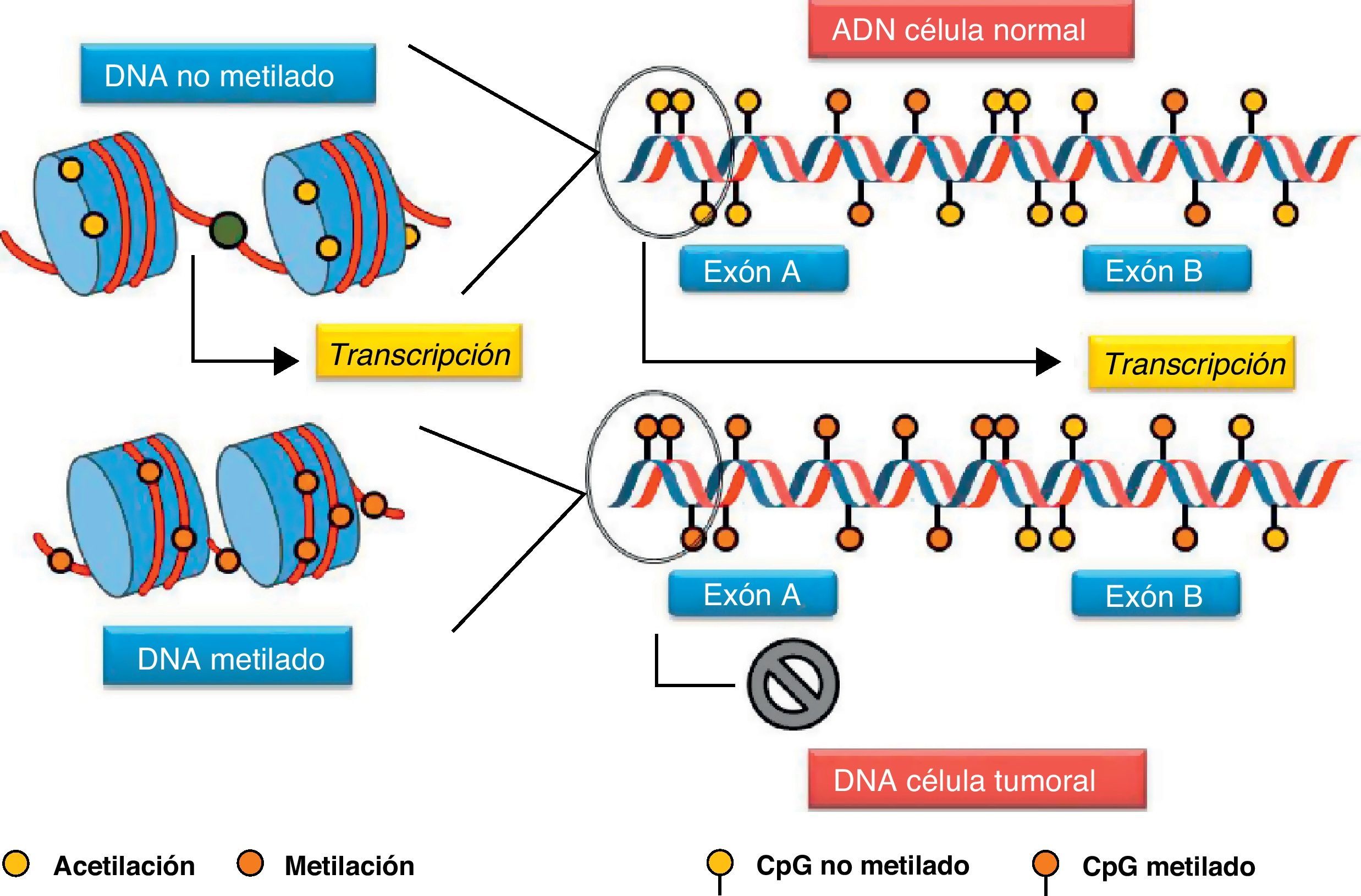
Aunque la metilación de los dinucleótidos CpG desempeña un papel importante en la regulación genética, también lo es la hipermetilación aberrante de los genes supresores de los tumores, los micro-ARN, los promotores… Característicamente, las CpG islands se mantienen desmetiladas durante el desarrollo y la diferenciación tisular, pero cuando se activan (…como en el caso de los genes supresores de tumores) se produce la acetilación y metilación de las histonas, lo que permite la transcripción de la cromatina abierta (fig. 3).

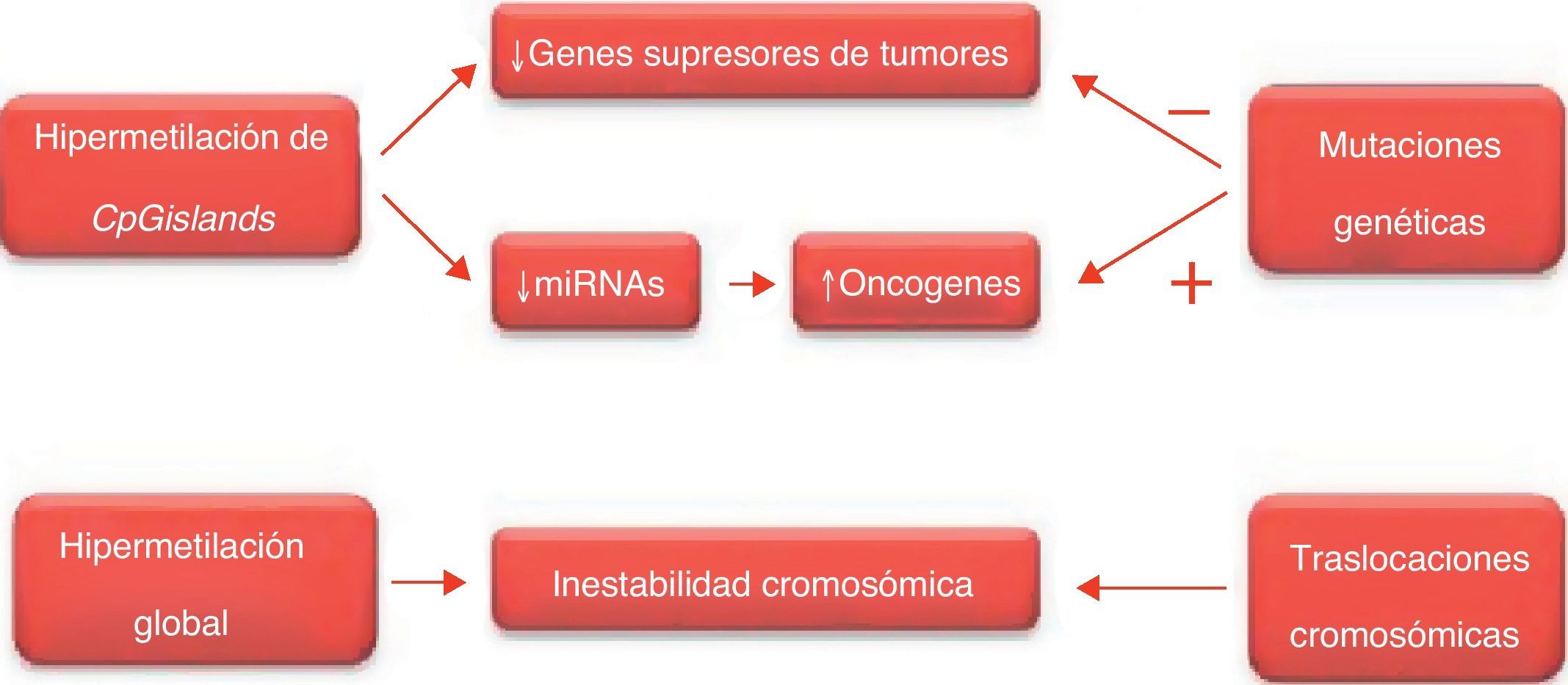
Durante la carcinogénesis, los promotores de los genes supresores de tumores con CpG islands son inusualmente metilados, no permitiéndose la apertura de la cromatina con el resultado de un silenciamiento aberrante2,3. Como consecuencia, la disminución de la actividad o el silenciamiento de los genes supresores de tumores son variables añadidas a la carcinogénesis, estableciéndose una regulación epigenética como clave del mecanismo en la promoción y desarrollo de la enfermedad neoplásica18. Además de la inactivación directa de los genes supresores de tumores, la hipermetilación del ADN puede ejercer su función indirectamente mediante el silenciamiento de los genes que codifican los factores de transcripción y los genes involucrados en la reparación del ADN; como en el caso del gen 06-metilguanina-ADN-metiltransferasa, previsor de la transición de una guanina por una adenina, que desencadenará la subsiguiente acumulación de este tipo de transiciones en genes reguladores como el KRAS y TP5319. De esta forma, las células acumulan lesiones genéticas que permiten el rápido desarrollo del cáncer por la participación de estos genes en múltiples vías del desarrollo celular (fig. 4)2,15.

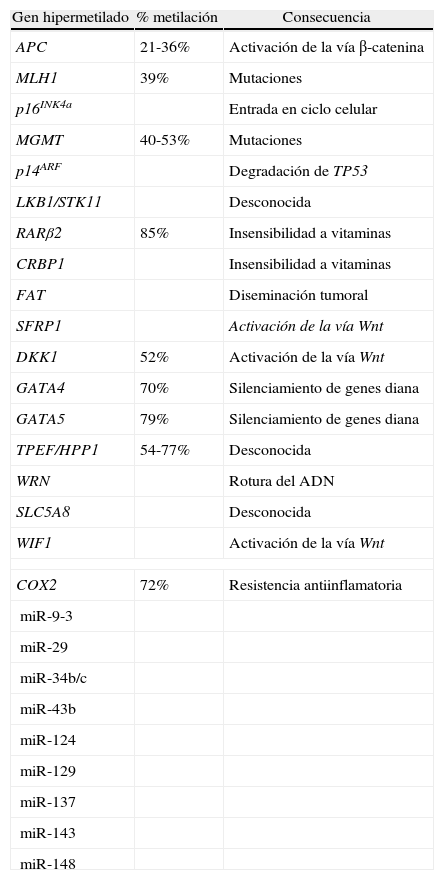
En algunas formas esporádicas de cáncer de colon existe un silenciamiento epigenético del gen STK11 (seronina/treonina kinasa 11), el cual regula la polaridad celular y actúa como gen supresor de tumores. Del 10-15% de los pacientes con cáncer de colon que presentan inestabilidad de los microsatélites, aproximadamente un 70-80% muestran silenciamiento epigenético de MLH1, un gen que procura reparar los errores por incompatibilidad entre bases20,21.El gen MLH1 pertenece al sistema de reparación del ADN, dando lugar a un silenciamiento transcripcional en situaciones de hipermetilación a nivel del promotor de dicho gen. Esta alteración epigenética se ha asociado a diferentes tipos de cáncer colorrectal esporádicos denominados fenotipo metilador de islas CpG (CIMP+). La vía carcinogenética de la inestabilidad de los microsatélites (MSI) también está relacionada con la mutación de los genes reparadores del ADN, siendo responsable del 15% del cáncer colorrectal esporádico. La clasificación molecular de los tumores colorrectales en función de la presencia o ausencia de CIMP y/o MSI es: CIMP+/MSI+ (frecuencia de 10%), CIMP+/MSI− (5-10%), CIMP−/MSI+ (5%) y CIMP−/MSI− (75-80%). Los carcinomas CIMP+, respecto a los CIMP−, suelen presentar una peor diferenciación, peor supervivencia, alta frecuencia de mutación del proto-oncogen BRAF y baja frecuencia de mutación del proto-oncogen KRAS; los marcadores más empleados en la determinación de este tipo de neoplasias son CDKN2A, MINT1, MINT2, MINT31 y MLH15,22. Los carcinomas MSI+, a diferencia de los CIMP+, se caracterizan por abundante secreción de mucina, infiltración linfocítica, localización en colon proximal, un menor estadio tumoral, mayor tamaño tumoral, menor frecuencia de metástasis ganglionares y hepáticas, menor frecuencia de mutaciones KRAS y, un mejor pronóstico22–24. Se han ejemplificado diferentes metilaciones relacionadas con el cáncer colorrectal; se aleja de los objetivos de esta revisión una puesta al día del resto de metilaciones existentes en la neoplasia colorrectal como puede ser la hipermetilación de genes como p16INK4a, CDKN2B/p15INK2B, MGMT, MINT1, MINT2, MINT31 y p14ARF (tabla 1)5,25–33.
Genes hipermetilados en el cáncer colorrectal y su consecuencia patológica
| Gen hipermetilado | % metilación | Consecuencia |
| APC | 21-36% | Activación de la vía β-catenina |
| MLH1 | 39% | Mutaciones |
| p16INK4a | Entrada en ciclo celular | |
| MGMT | 40-53% | Mutaciones |
| p14ARF | Degradación de TP53 | |
| LKB1/STK11 | Desconocida | |
| RARβ2 | 85% | Insensibilidad a vitaminas |
| CRBP1 | Insensibilidad a vitaminas | |
| FAT | Diseminación tumoral | |
| SFRP1 | Activación de la vía Wnt | |
| DKK1 | 52% | Activación de la vía Wnt |
| GATA4 | 70% | Silenciamiento de genes diana |
| GATA5 | 79% | Silenciamiento de genes diana |
| TPEF/HPP1 | 54-77% | Desconocida |
| WRN | Rotura del ADN | |
| SLC5A8 | Desconocida | |
| WIF1 | Activación de la vía Wnt | |
| COX2 | 72% | Resistencia antiinflamatoria |
| miR-9-3 | ||
| miR-29 | ||
| miR-34b/c | ||
| miR-43b | ||
| miR-124 | ||
| miR-129 | ||
| miR-137 | ||
| miR-143 | ||
| miR-148 | ||
Fuente (basada): 5,22,24,34,37–39.
Gran parte del código epigenético es transportado, a través de las modificaciones químicas post-transcripcionales, a nivel de los aminoácidos de las histonas4,15. Las histonas constan de un dominio globular C-terminal y otro no estructurado N-terminal, los cuales pueden sufrir modificaciones covalentes tras una traducción del tipo metilación, acetilación, fosforilación o ubiquitinización. Estas circunstancias van a determinar la regulación de los procesos celulares de la transcripción, la replicación, la reparación del ADN y la organización cromosómica, mediante la conformación de la estructura y la actividad de la cromatina. Estas modificaciones reversibles favorecen bien la forma inactiva de la cromatina (heterocromatina) o bien su configuración activa (eucromatina)2,4,15.
El proceso de modificación de las histonas se lleva a cabo a través de diferentes grupos de enzimas (acetiltransferasas, desacetilasas, metiltransferasas) que incorporan o eliminan de una forma dinámica las modificaciones covalentes, regulando así la activación de la cromatina2,15. Estas variaciones en la cromatina incluyen: la pérdida de acetilación de las histonas, que implica la represión genética; la sobreexpresión de las desacetilasas de las histonas, presentes en diversos tipos tumorales; la alteración de la metilación, asociada con el silenciamiento aberrante de los genes supresores de tumores; la modificación de la expresión de las demetilasas, implicadas en la progresión tumoral2.
La activación o inhibición de la cromatina depende de los residuos modificados, con el resultado de un silenciamiento genético a través de la formación de una estructura de cromatina compacta. La acetilación de la lisina se relaciona con la activación de la transcripción, mientras que su metilación conlleva una activación o inhibición en función del tipo de metilación: la metilación de H3 en K4 implica una activación de la transcripción, mientras que la metilación de H3 en K9 o en K27 y la metilación de H4 en K20 conlleva una inhibición de la transcripción. A su vez, estos cambios se ven influenciados por la metilación de la secuencia del ADN en los CpG islands2,15,16.
Las desacetilasas de las histonas ayudan a controlar la estabilidad proteica, ya que las lisinas también son señalizadas por ubiquitinización para luego ser degradadas, por la vía del proteasoma, reduciéndose así la vida media de la proteína. La acetilación participa igualmente en la importación y exportación de las proteínas en la membrana nuclear merced a las importinas; además de en otros procesos como la formación de microtúbulos, el control del estrés interno y externo, así como participación en las vías de inflamación, apoptosis y necrosis2,21.
En el cáncer colorrectal se han determinado varias alteraciones genéticas a nivel de diferentes acetiltransferasas como CBP, pCAF o p300, cuya principal función es la regulación de la transcripción. Mientras que CBP puede presentar mutaciones, deleciones y traslocaciones, únicamente se han determinado mutaciones en pCAF y p300, caracterizándose esta última por presentarse en tumores que muestran inestabilidad de los microsatélites25,26,30,31.
Cambios inducidos por ácido ribonucleico no codificantesExiste una evidencia creciente de la relación existente entre los micro-ARN y el cáncer colorrectal. Los micro-ARN son estructuras moleculares de 20-22 nucleótidos con actividad post-transcripcional a través de la unión con el m-RNA; están implicados en la regulación de la expresión genética al estar involucrados en la apoptosis, la proliferación y la diferenciación celulares, habiéndose demostrado su funcionalidad como genes supresores de tumores o como proto-oncogenes en la carcinogénesis. Más del 50% de los genes de los micro-ARN están localizados en regiones frágiles, en regiones mínimas de amplificación o de pérdida de heterocigosidad y en regiones de puntos de rotura, frecuentemente relacionados con las neoplasias34,35.
Los cambios producidos en los ARN no codificantes, incluidos los micro-ARN, junto con la metilación del ADN y las modificaciones de las histonas, forman parte de los cambios heredables en el ámbito de la epigenética, por regular las enzimas responsables de la metilación del ADN (DNMT3A y DNMT3B) y las modificaciones de las histonas (EZH2). Además se ha sugerido que la expresión de los micro-ARN está a su vez regulada por dichas modificaciones epigenéticas, hallándose aproximadamente el 10% de los micro-ARN regulados por la metilación del ADN2.
La primera referencia que ponía de manifiesto la implicación de los micro-ARN en la epigenética del cáncer fue la demostración del silenciamiento del micro-ARN supresor de tumores miR-127 por la hipermetilación de los islotes CpG del ADN1. Uno de los hallazgos más importantes recientemente comunicado es la regulación de los micro-ARN por el TP53, al constatar que la familia miR-34 está directamente regulada e involucrada en la función antitumoral del TP53. En el cáncer colorrectal, la transcripción de la región CpG-miR-34b/c podría estar estimulada por el TP53, existiendo «potenciales zonas ligando» de este supresor de tumores en aproximadamente el 46% de las regiones promotoras de los micro-ARN, lo que sugeriría la regulación de los micro-ARN por parte del TP5328. De la misma forma, la activación de Myc implica una supresión de la expresión de los micro-ARN, como sería el caso del cluster miR-17-9236.Junto a la regulación de los micro-ARN por parte del TP53, se ha demostrado el silenciamiento de miR-342 en el cáncer colorrectal a través de la metilación de los islotes CpG del gen evl. Asimismo, la hipermetilación de islotes CpG de diferentes tipos celulares de neoplasias supone la inactivación del miR-124a. En el cáncer colorrectal se han determinado también otros micro-ARN que presentan una alteración epigenética como son miR-9, miR-43b, miR-34c, miR-124a, miR-129, miR-137, miR-143, miR-148 o miR-34234,37–39.
Otra estrategia que ha permitido demostrar la regulación epigenética de los micro-ARN, es el uso de los inhibidores de las ADN metiltransferasas y de los inhibidores de las desacetilasas de las histonas. Se ha conseguido identificar varios micro-ARN, regulados por la hipermetilación de las regiones CpG de miR-9-1, miR-129-2 y miR-137 en el cáncer colorrectal, asociándose la metilación de miR-9-1 a la presencia de metástasis linfáticas35,40,41.
Los micro-ARN participan en la epigenética del cáncer tanto por estar regulados por la metilación del ADN y por la modificación de las histonas, como por colaborar en la regulación de dichos procesos epigenéticos, ya que las ADN metiltransferasas, las deacetilasas de las histonas y las metiltransferasas de las histonas son dianas moleculares de los micro-ARN. Así se ha demostrado, por ejemplo, que el miR-29b induce una hipometilación del ADN por disminución de la expresión de las ADN metiltransferasas, mediante la interacción directa con DNMT3A y DNMT3B e indirectamente con DNMT1. Considerando que DNMT3A es también una diana molecular de miR-143, un micro-ARN frecuentemente regulado a la baja en el cáncer colorrectal36.
Aplicaciones clínicas de los marcadores epigenéticosLos cambios en el estado de metilación del ADN y la modificación de la cromatina son características de las células tumorales, por lo que sus patrones diferenciales podrían ser útiles en el diagnóstico y clasificación de la enfermedad neoplásica. La PCR en tiempo real es una técnica de determinación de la metilación con alta sensibilidad y especificidad. El ADN es una molécula estable que se puede obtener de múltiples fuentes y que puede emplearse tiempo después de su extracción5,42,43.
Las diferentes fuentes de obtención del ADN (células descamadas, orina, plasma, saliva, lavado bronquial…) permitirían la determinación de los patrones de hipermetilación de los islotes CpG, favoreciéndose el diagnóstico tumoral temprano especialmente en pacientes con historia de cáncer familiar. Tras el aislamiento del ADN, las muestras son tratadas con bisulfito de sodio, transformándose las citosinas no metiladas en uracilos, mientras que las citosinas metiladas permanecen intactas, determinándose así la metilación aberrante del ADN con la PCR a tiempo real. Mediante esta técnica se han determinado promotores frecuentemente metilados de forma aberrante en estadios precoces de cáncer colorrectal como ALX4, SEPT9 y TMEFF25,41,44.
Tras el diagnóstico de la neoplasia es importante establecer qué tratamiento resultará el más adecuado, siendo preciso conocer los genes hipermetilados que participan en el desarrollo de la enfermedad. Así, la eficiencia de la reparación del ADN dañado constituirá un componente importante en la resistencia al tratamiento. Ejemplo de lo cual sería la participación de la hipermetilación del promotor del gen MLH1, puesto que la desmetilación del promotor induce una mayor sensibilización frente al tratamiento con fármacos quimioterápicos como el cisplatino. Otros estudios han postulado que la activación del mi-ARN supresor de tumores miR-127 por drogas modificadoras de la cromatina, podría inhibir el crecimiento tumoral a través de la regulación «a la baja» de sus oncogenes diana5,45,46. Conocidas las implicaciones de los micro-ARN en el proceso neoplásico y su participación en el código epigenético, diversos estudios se han orientado a la conjugación de la epigenética y de los micro-ARN en el tratamiento del proceso tumoral. La característica reversibilidad de los procesos epigenéticos, como la metilación del ADN y la acetilación de las histonas, ha hecho que se generen nuevas perspectivas terapéuticas tales como la 5-aza-2′-deoxicitidina y la 5-azacitidina, utilizados como inhibidores de la metilación del ADN16,42.
ConclusionesLa observación de las células tumorales ha permitido establecer que en el origen de la enfermedad neoplásica las alteraciones epigenéticas pueden ser tan importantes como las modificaciones genéticas. Ambas alteraciones contribuyen a la progresión y desarrollo de la enfermedad neoplásica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

