




Introducción
La pancreatitis hereditaria es una enfermedad autosómica dominante caracterizada clínicamente por múltiples episodios recurrentes de pancreatitis aguda, normalmente iniciados a edades muy tempranas que, finalmente, dan lugar al desarrollo de una pancreatitis crónica1-3. En este grupo de pacientes se ha visto, además, que el riesgo de padecer cáncer de páncreas se estima en más de 50 veces el normal. En la mayoría de los casos es causada por mutaciones en el gen del tripsinógeno catiónico (PRSS1), normalmente R122H o N29I1-3. A pesar de los avances sucedidos en el campo diagnóstico, la mayoría de estos pacientes quedan sin un diagnóstico etiológico final, y se considera que presentan pancreatitis agudas de repetición de carácter idiopático1-3.
Presentamos un caso de un paciente con pancreatitis agudas de repetición, inicialmente etiquetadas como de origen idiopático, al que, tras la secuenciación del gen del tripsinógeno, se le detectó una mutación sólo descrita con anterioridad en 1 familia4.
Caso clínico
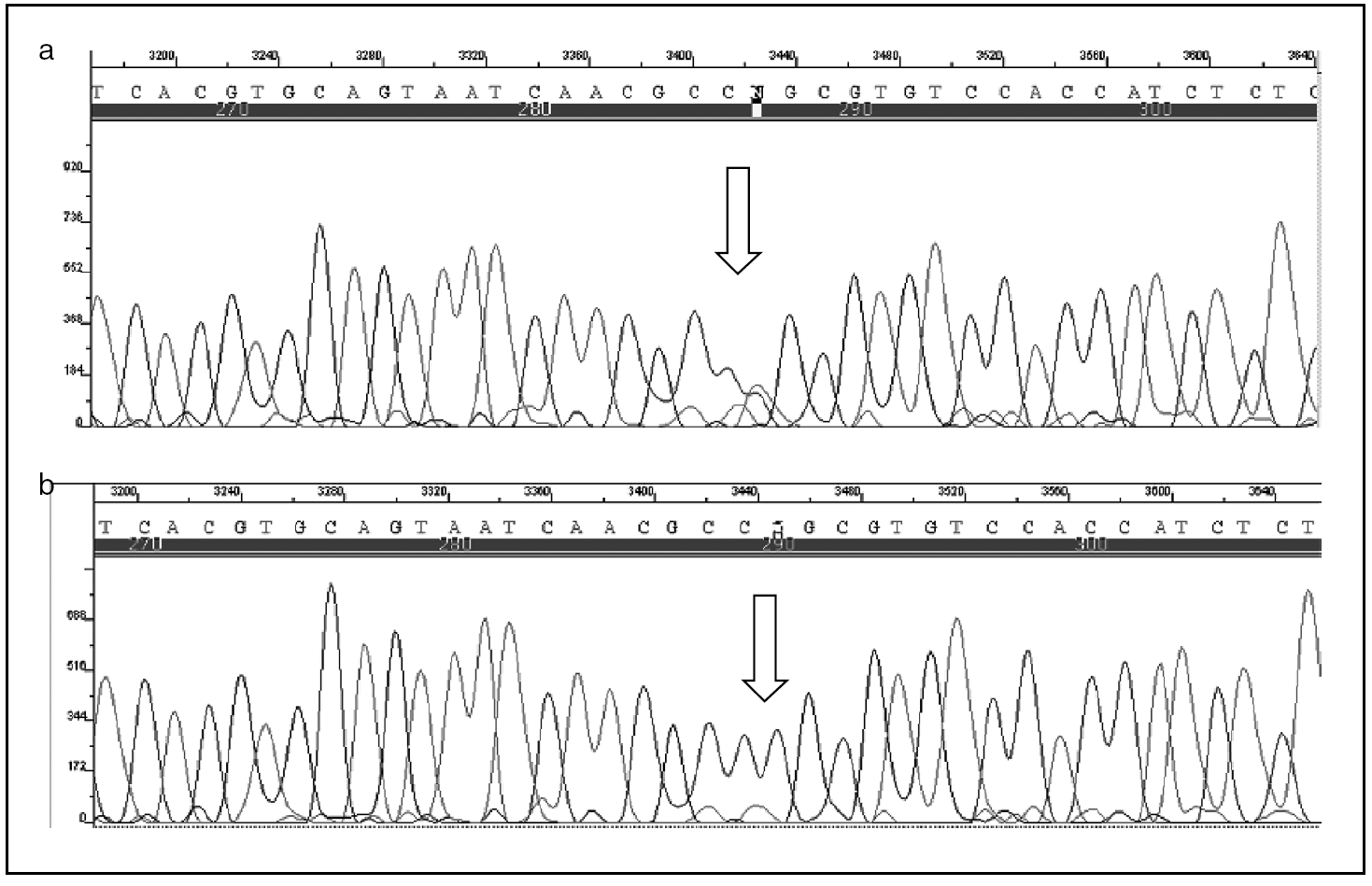
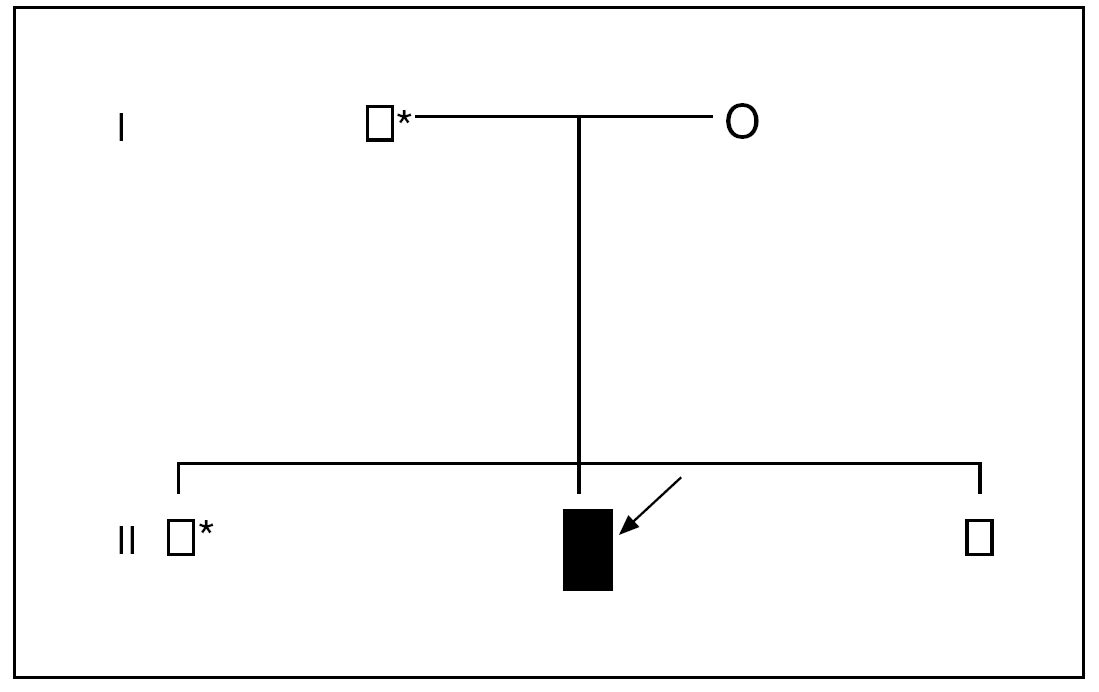
Varón de 12 años, sin antecedentes personales o familiares de interés, en cuya historia clínica destaca haber padecido, desde los 8 años, 10 episodios de pancreatitis agudas, todas ellas de carácter leve, de las que 5 precisaron de ingreso hospitalario. Las pancreatitis se etiquetaron, en todas las ocasiones, como de origen idiopático, una vez se completaron las siguientes pruebas complementarias: estudio analítico completo, con especial atención a la calcemia, el lipidograma, etc., ecografía abdominal, colangiopancreatografía retrógrada endoscópica, tomografía computarizada (TC), resonancia magnética (RM), colangio-RM y ecoendoscopia. Ante la persistencia de los episodios de pancreatitis y la ausencia de un diagnóstico etiológico se decidió investigar la posibilidad de mutaciones en el gen del tripsinógeno catiónico (PRSS1); se estudiaron todos los exones y las regiones adyacentes de dicho gen mediante técnicas de reacción en cadena de la polimerasa (PCR) y secuenciación. Se identificó, en la línea germinal, el cambio del nucleótido citosina (C) por timina (T) en la posición 364 de la secuencia de uno de los alelos del gen estudiado en el exón 3 (PRSS1: c.364C > T), lo que supone la sustitución del aminoácido arginina por cisteína en posición 122 (fig. 1). Ante estos hallazgos, se efectuó idéntico análisis genético al resto de la familia, lo que permitió identificar a un hermano y al padre como portadores sanos (fig. 2).
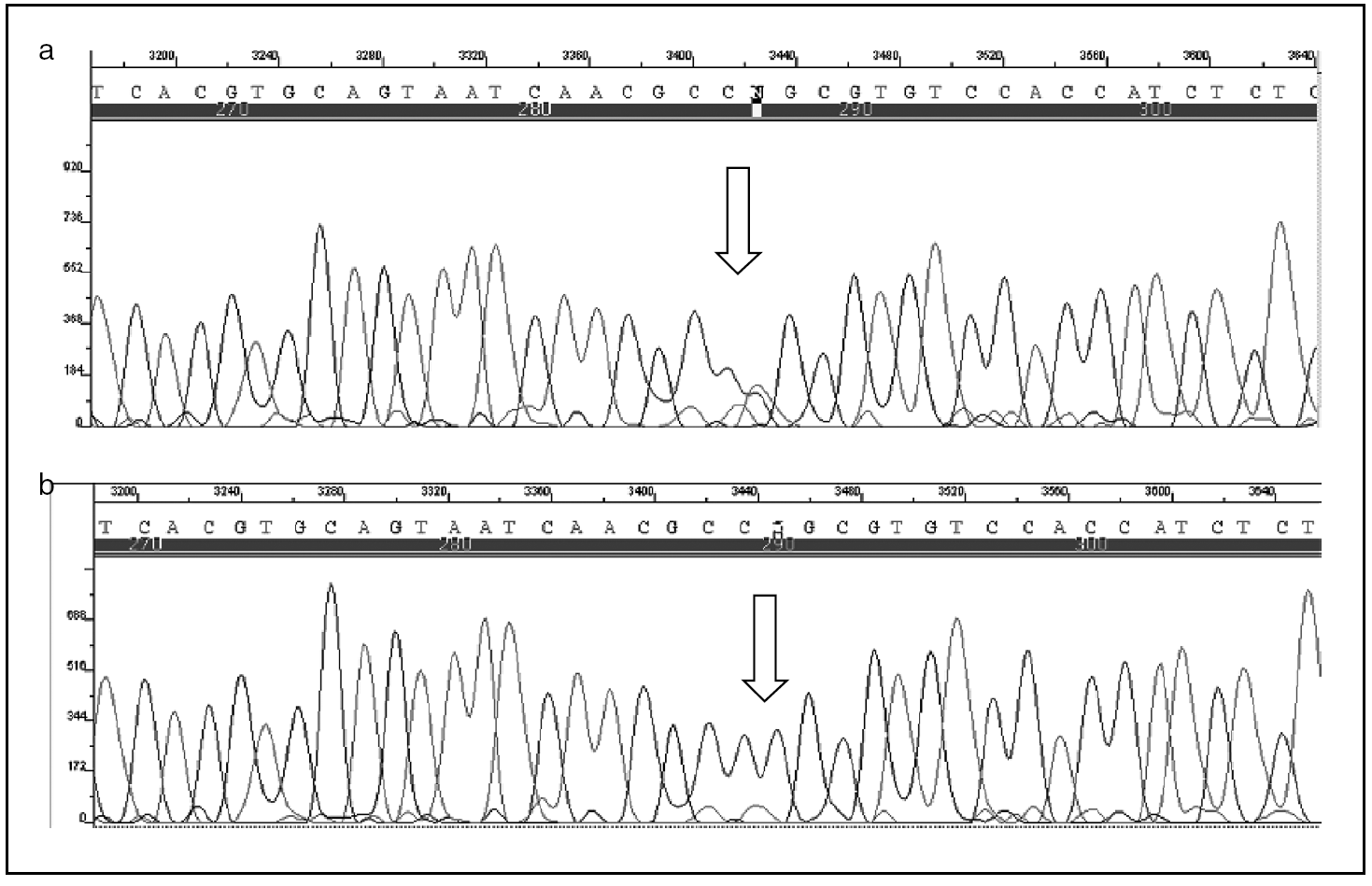
Fig. 1. Análisis de la secuencia del exón 3 del gen del tripsinógeno catiónico. La imagen 1a corresponde a un paciente control, y se observa, en comparación con la imagen 1b, perteneciente al caso descrito, cómo en posición 364 la citosina ha sido sustituida por timina.
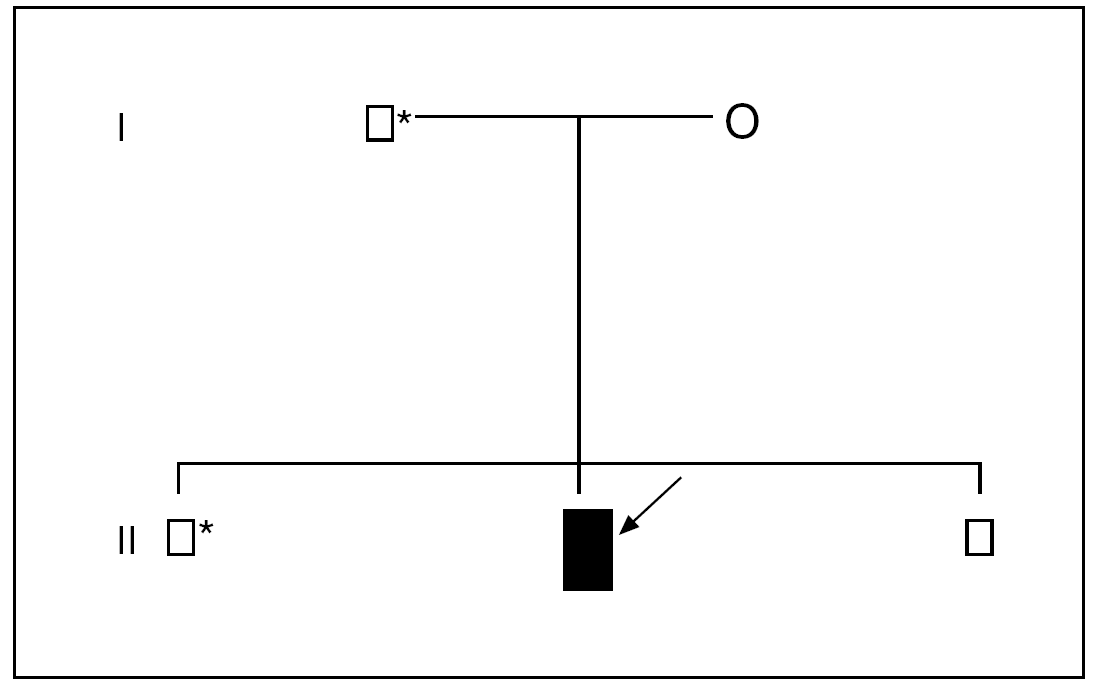
Fig. 2. Genograma de la familia afectada. Varón sano; varón enfermo; * varón portador; O mujer sana. La flecha señala el caso índice.
Discusión
La pancreatitis hereditaria (PH) es una rara variedad de pancreatitis, heredada de forma autosómica dominante con una penetrancia del 80%, y que se caracteriza clínicamente por la presencia de múltiples y recurrentes episodios de pancreatitis aguda. Los síntomas suelen iniciarse en la infancia, y con el tiempo estos pacientes acaban desarrollando una pancreatitis crónica1-3. Estos pacientes presentan, además, un riesgo muy elevado de presentar cáncer de páncreas, y se calcula que hasta un 40% de los pacientes lo desarrollará a lo largo de su vida5.
El primer informe de PH fue publicado por Comfort y Steinberg, de la Mayo Clinic, en 19526. Posteriormente se identificaron muchas otras genealogías en todo el mundo que sirvieron como base para los estudios genéticos moleculares. En mayo de 1966, Whitcomb et al7 identificaron la primera mutación especifica de la enfermedad, la R122H. En ésta, que es la más frecuente (el 80-90% de los casos), existe un cambio del nucleótido guadinina (G) por adenosina (A) de la secuencia de uno de los alelos del gen del tripsinógeno catiónico en el exón 3 (PRSS1: cG > A), lo que supone la sustitución del aminoácido arginina por histidina en la posición 122 (R122H). Un año más tarde, se descubre otra mutación específica, la N29I8. Hasta la fecha, se han identificado cerca de 10 diferentes mutaciones del PRSS1 asociadas a PH8,9. La mutación que presenta la familia aquí descrita (R122C) sólo se ha identificado con anterioridad en 1 familia4.
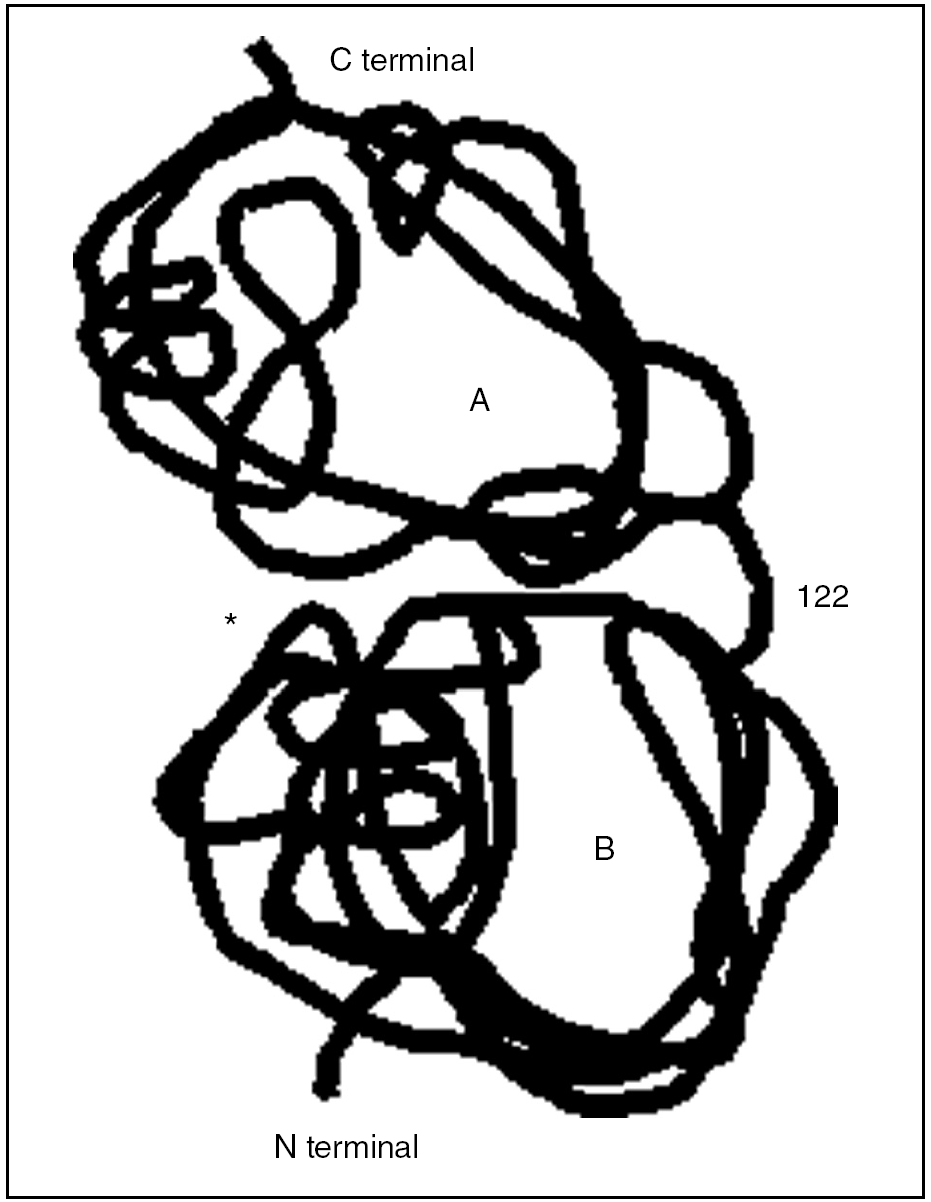
La clave fisiopatológica del desarrollo de pancreatitis agudas en pacientes con PH reside en que la mutación en el gen del PRSS1 hace que falle el mecanismo de inactivación de la tripsina catiónica activada de forma prematura (fig. 3). En condiciones normales, la tripsina activada intracelularmente es inactivada por autodigestión, al fraccionarse la molécula en 2 dominios en la posición 122 en la arginina. En el caso de la mutación R122H, o la aquí descrita (R122C), se elimina el sitio de autólisis de la tripsina, lo que crea una "supertripsina" que no puede ser inactivada7-9.
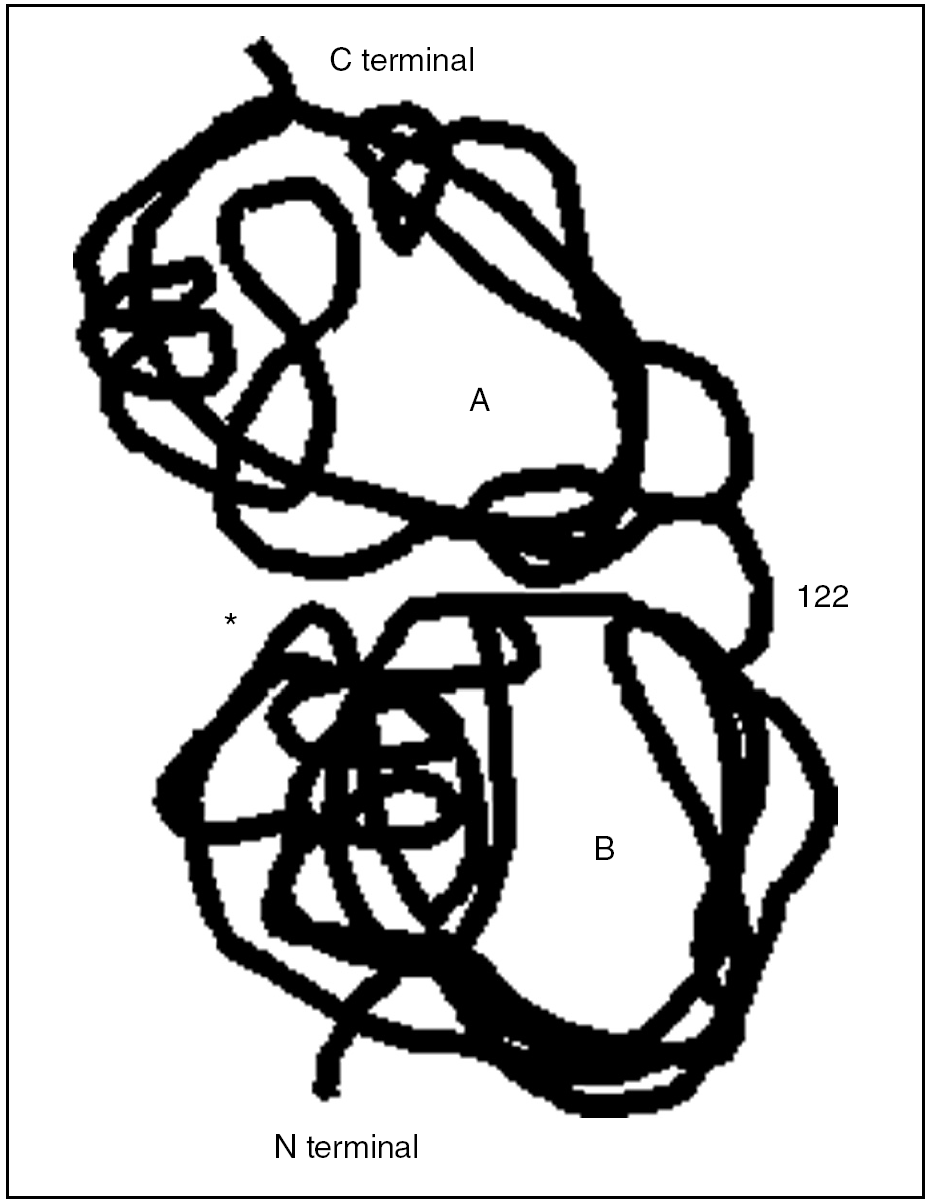
Fig. 3. Se representa la estructura molecular del tripsinógeno catiónico con sus 2 dominios moleculares, A y B. *Bolsillo de especificidad o sitio activo de la enzima. La posición 122 corresponde a la zona de unión de ambos dominios, a su vez, el punto de corte y la acción de la tripsina (autoinactivación).
Esta enfermedad, si bien poco frecuente, es diagnosticada cada vez con mayor frecuencia, especialmente en el grupo de pacientes inicialmente etiquetados como de origen idiopático. Siguiendo las recomendaciones de la conferencia de consenso sobre tests genéticos en PH celebrada en el tercer Simposium Internacional en Enfermedades Hereditarias del Páncreas10, las indicaciones para buscar mutaciones del gen del PRSS1 serían cualquiera de las siguientes: a) 2 o más episodios de pancreatitis aguda idiopática recurrente; b) pancreatitis crónica idiopática; c) historia familiar de pancratitis en familiares de primer o segundo grado; d) un episodio de pancreatitis aguda que requiere hospitalización en un niño, y e) pacientes con pancreatitis aguda que cumplen criterios de estudio según el comité ético local. No existen tratamientos médicos específicos en la PH. El tratamiento de los episodios agudos debe ser idéntico al empleado en pancreatitis de cualquier otro origen. Asimismo, la pancreatitis crónica, una vez desarrollada no posee, tampoco, tratamiento específico alguno1-3.
Desde el punto de vista del consejo genético, debe informarse a los familiares que tanto los portadores sanos como los individuos pertenecientes a estas familias, pero que no poseen la mutación, al no desarrollar episodios de pancreatitis agudas, no poseen riesgo elevado de presentar cáncer de páncreas, por lo que en ellos no estaría indicada ninguna medida de cribado. Dado que los pacientes con PH poseen un riesgo de desarrollar un cáncer de páncreas 53 veces el normal, con un riesgo acumulado de padecerlo en la vida del 40%, su cribado y su diagnóstico temprano parece esencial. Los marcadores tumorales y las técnicas de imagen y endoscopia poseen una escasa sensibilidad y especificidad en el diagnóstico temprano. Más aún, la detección de tumores es particularmente difícil en el contexto de pancreatitis crónica. El protocolo utilizado en el programa EUROPAC1 para pacientes PH de más de 30 años se basa en la utilización conjunta de TC más ecoendoscopia con toma de muestras de jugo pancreático para análisis de mutaciones K-ras. La pancreatectomía total debe de ser la última medida que emplear, y se recomienda sólo en caso de sospecha elevada de cáncer (presencia de atipias celulares o anormalidades focales) y en los pacientes que vayan a ser sometidos a cirugía pancreática por pancreatitis crónica. Su empleo fuera de estas indicaciones está totalmente contraindicado.
En resumen, la PH es una rara enfermedad caracterizada por episodios recurrentes de pancreatitis aguda que, a diferencia de las producidas en el seno de otras enfermedades comienza con la activación de la tripsina a escala intracelular por fallo en los mecanismos de autoinactivación, normalmente por mutaciones en del gen del tripsinógeno catiónico. Estos pacientes, que poseen un riesgo de cáncer de páncreas muy aumentado, con frecuencia acaban desarrollando una pancreatitis crónica, pero no por obstrucción ductal crónica, si no secundaria a los múltiples episodios de pancreatitis sufridos.
Correspondencia: Dr. J.A. Fernández.
Servico de Cirugía I, 3.a planta. Hospital Virgen de la Arrixaca.
Murcia-Cartagena, s/n. 30120 Murcia. España.
Manuscrito recibido el 20-12-2004 y aceptado el 5-9-2005.

