




Mujer de 48 años sin antecedentes familiares conocidos de síndrome MEN 2A, que es intervenida en abril de 1989 de un nódulo tiroideo. Se practicó tiroidectomía total y la anatomía patológica fue de correspondió a cáncer medular de tiroides (CMT) bilateral. Ante la posibilidad de que la paciente fuese portadora de una mutación RET, se solicitó estudio genético, TC suprarrenal, catecolaminas en orina 24h y estudio paratiroideo. El calcio, fósforo y PTH fueron normales. Se detectó un leve aumento de metanefrinas en orina de 24h (metanefrinas 640mcg/24h)(valores normales 60-350mcg/24h) y la TC suprarrenal demostró la existencia de ambas suprarrenales tumorales (1,5cm la derecha y 2,5cm la izquierda). La genética confirmó una mutación RET: c.1901G>A (p.C634Y). Con estos hallazgos se procedió a realizar una suprarrenalectomía bilateral.
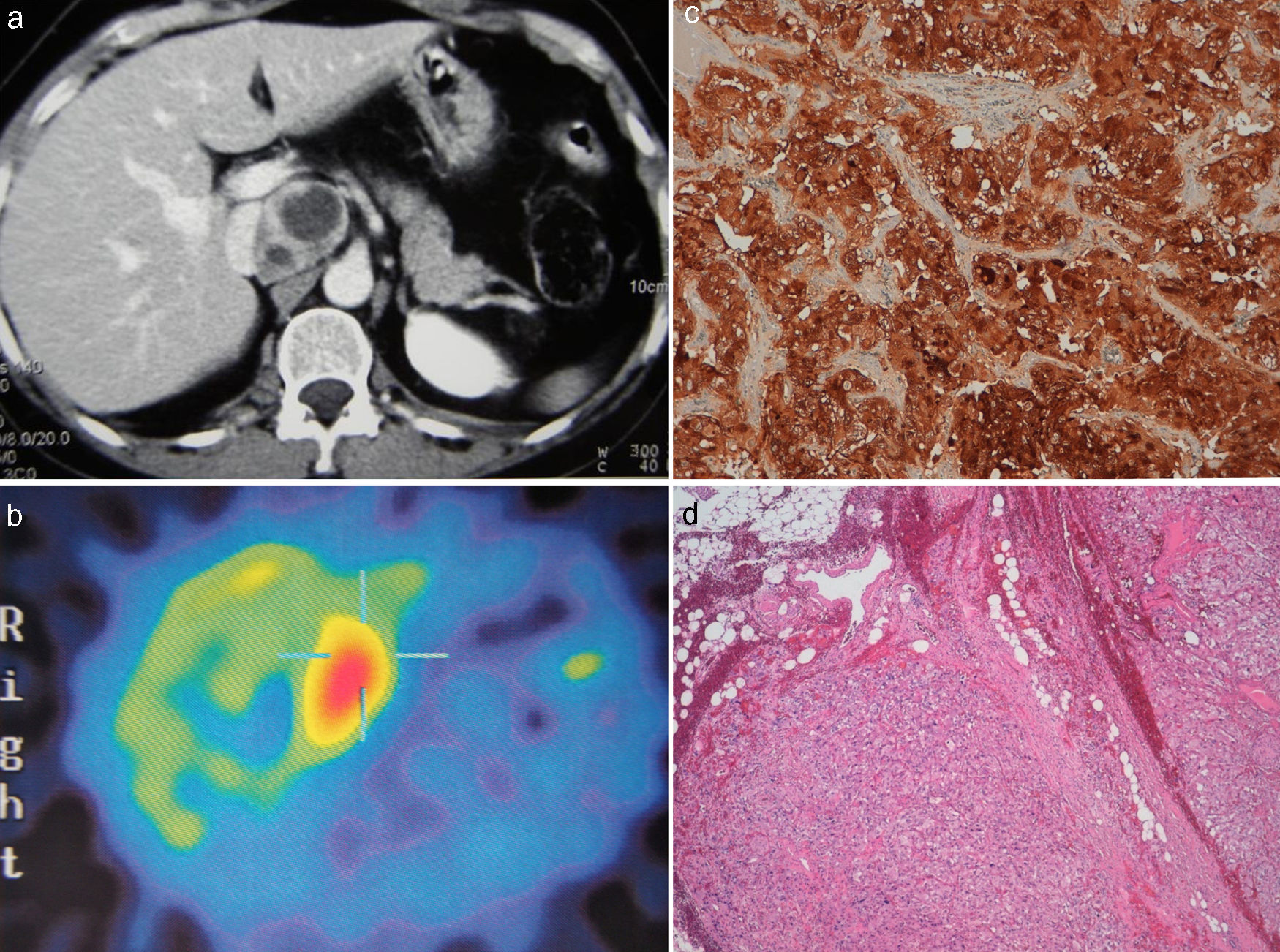
Tras 15 años de seguimiento sin incidencias, se detectó nueva elevación de los niveles de presión arterial junto a un incremento de los valores de catecolaminas en orina 24h (noradrenalina, 140mcg/24h (vn 12-86); adrenalina, 150mcg/24h (vn 2-23); normetanefrina, 684mcg/24h (vn 120-650); metanefrinas totales, 2.444mcg/24h (vn 180-1000); catecolaminas totales, 289mcg/24h (vn 14-110); AVM, 25,6mg/24h (vn 1-10). La TC abdominal evidenció una masa de centro hipodenso de unos 4,5×3cm en el espacio interaortocava por encima de la salida de la vena renal izquierda y en íntimo contacto con la vena cava (fig. 1a). Se realizó una gammagrafía con metayodobencilguanidina (MIBG), cuyos hallazgos fueron compatibles con masa hipercaptante en posición suprarrenal derecha sugestiva de recidiva de feocromocitoma derecho o paraganglioma (fig. 1b).

a: TC abdominal: masa de centro hipodenso de unos 4,5 x 3 cms. en el espacio interaortocava por encima de la salida de la vena renal izquierda y en íntimo contacto con la vena cava. b: MIBG: masa hipercaptante en posición suprarrenal derecha sugestiva de recidiva de feocromocitoma derecho o paraganglioma. c: Inmunohistoquímica: positividad inmunohistoquímica para cromogranina A, sinaptofisina, enolasa y vimentina. d: Anatomía Patológica: tumoración con superficie limitada por una pseudocápsula fibrosa evidenciando en su interior células ganglionares.
La paciente, previa preparación con fenoxibenzamina, se intervino quirúrgicamente, evidenciando una tumoración de unos 4-5cm. alojada en la celda formada por la vena cava, la vena renal izquierda y la aorta, y se procedió a su exéresis completa. El postoperatorio cursó sin complicaciones, normalizándose las cifras de catecolaminas. El estudio anatomopatológico e inmunohistoquímico de la pieza se informó como tumoración de 5.5×4,5×2,5cm de superficie limitada por una pseudocápsula fibrosa evidenciando en su interior células ganglionares con positividad inmunohistoquímica para cromogranina A, sinaptofisina, enolasa y vimentina, compatible con paraganglioma (fig. 1c-d). Tras la última intervención, la paciente se mantuvo clínicamente asitomática con cifras de catecolaminas en orina 24h normales.
El MEN 2A es una enfermedad poco frecuente que asocia CMT en el 100% de los casos, feocromocitoma en el 50% e hiperparatiroidismo en el 10-15% de los casos1. La asociación de paragangliomas con MEN 2A es excepcional como así lo refiere la literatura científica descrita hasta el momento2 (búsqueda bibliográfica en medline a fecha de febrero de 2011).
Los feocromocitomas y los paragangliomas extraadrenales son tumores de estirpe cromafin que se localizan en el 95% de los casos a nivel intraabdominal (fundamentalmente adrenales), el 2-4% a nivel torácico y el 1% en cuello. Los paragangliomas se suelen localizar a nivel de la cadena simpática ganglionar, siendo los retroperitoneales extraadrenales los de peor pronóstico2. El 20% de estos paragangliomas tienen potencial maligno, suelen ser multicéntricos caracterizándose por la recurrencia local o aparición de metástasis en un gran número de ocasiones. Esta recurrencia puede aparecer tras años o décadas de la exéresis del tumor primario por lo que el seguimiento a largo plazo es necesario3.
Las manifestaciones clínicas están determinadas por la capacidad que tienen algunos de ellos (39%) para secretar hormonas como catecolaminas, gastrina, tirocalcitonina, ACTH, VIP y PTH, entre otras, siendo la hipertensión arterial el síntoma más común3. En el caso que describimos, la elevación de los niveles hormonales y de las cifras de presión arterial condujeron a la sospecha diagnóstica.
El diagnóstico de feocromocitomas y paragnagliomas se realiza mediante una buena anamnesis y exploración física junto a la determinación de los niveles de catecolaminas plasmáticas y/o metanefrinas en orina de 24h. La localización del tumor puede realizarse con técnicas como la MIBG, que tiene capacidad para detectar tumores <0,5cm, metastásicos y multicéntricos, la TC, RMN o PET. Estas técnicas también son utilizadas para el seguimiento de estos pacientes para el diagnóstico temprano de posibles recidivas. En el caso que presentamos la MIBG y la TC detectaron la existencia del tumor, siendo el diagnóstico histológico e inmunohistoquímico el que realizó el diagnóstico definitivo4.
En cuanto al tratamiento del feocromocitoma, la suprarrenalectomía uni- o bilateral es la técnica quirúrgica de elección, siendo el abordaje laparoscópico actualmente un procedimiento seguro y con buenos resultados. En caso de paraganglioma, la exéresis quirúrgica es el tratamiento de elección, realizando una revisión completa de la cavidad abdominal y pélvica en busca de otros tumores. En el caso de nuestra paciente no se encontró ninguna otra afectación. En los feocromocitomas malignos o metastásicos se puede asociar tratamiento con MIBG-I131 a dosis altas (200mCi), pudiéndose repetir la dosis hasta un total de 800 a 1.200mCi. Se consigue mejoría del tamaño de las metástasis en un 50%, pero la remisión completa se alcanza solo en un 4% de los casos. El uso de quimioterapia y/o radioterapia no mejora los resultados de la cirugía y se utilizan como tratamiento paliativo en enfermos con enfermedad avanzada5.
En conclusión, cabe destacar que la asociación de paragangliomas con MEN 2 está descrita en la literatura científica como excepcional y que se puede confundir con el diagnóstico de feocromocitoma o con la ausencia de curación del mismo tras su exéresis, pudiendo aparecer incluso años más tarde, por lo que el seguimiento a largo plazo de estos pacientes es obligatorio por la posible aparición de recidivas, metástasis o, como en nuestro caso, de paraganglioma asociado, siendo la MIBG, junto con la determinación de catecolaminas, las pruebas de elección.

