





Sr. Director:
Los tumores neuroendocrinos del páncreas son una rara entidad con un amplio espectro en su presentación clínica. Aproximadamente el 50% son no funcionantes. De los funcionantes, el más frecuente es el insulinoma, seguido del gastrinoma; y en menor frecuencia aparecen los vipomas, glucagonomas y acthomas1,2.
Alrededor del 1% de los síndromes de Cushing son causados por los tumores pancreáticos productores de ACTH. Este tipo de tumor se localiza sobre todo en cuerpo y cola pancreáticos3.
Clínicamente asocian repentina ganancia de peso, con obesidad central, pérdida de masa muscular, intolerancia a la glucosa y aumento de la presión arterial.
El diagnóstico de la producción ectópica de ACTH se confirma con el fallo en la supresión urinaria de cortisol y 17-hidrocorticosteroide con altas dosis de dexometasona; ausencia de respuesta adrenal tras estimulación hipofisaria y aumento de ACTH plasmática sin gradiente positivo en los senos petrosos venosos. Para su localización la tomografía computarizada y la resonancia magnética son las pruebas indicadas.
Su tratamiento es quirúrgico aunque, dadas la agresividad y la progresión clínica, un alto porcentaje presenta metástasis hepáticas al diagnóstico. La cirugía debe realizarse, si es posible, incluyendo dichas metástasis. La adrenalectomía bilateral es una opción terapéutica que considerar en la cirugía3.
Se trata de un paciente varón que consulta por astenia, hinchazón de cara, cuello y nuca. Asocia disminución del vello en extremidades inferiores y pelo en cuero cabelludo, refiere disminución de masa muscular en extremidades inferiores y debilidad de predominio proximal.
Entre sus antecedentes destacan hipertensión y fibrilación auricular.
En la exploración física se objetiva un peso de 81 kg, una talla de 173 cm y 155/100 mmHg de presión arterial; llama la atención la acumulación de grasa en las zonas cervical y troncular.
Con la sospecha de síndrome de Cushing, se realizan determinaciones hormonales: TSH, T3 y T4 normales; cortisol plasmático, 24,5 mg/dl; ACTH, 124 pg/ml; cortisol libre en orina, 905 μg/24 h. El resto de las determinaciones fue normal.
Los test de frenado con 1 y 8 mg de dexametasona fueron negativos: cortisol plasmático de 28,9 y 25,4 μg/dl, respectivamente. A continuación se practicó una resonancia magnética hipofisaria, que objetivó hipocaptación en margen izquierda de adenohipófisis, y otra adrenal, que objetivó un pequeño nódulo de 1 cm en brazo medial de la suprarrenal izquierda, compatible con adenoma.
Ante los datos hormonales que indicaban un origen ectópico pero sin poderse descartar totalmente el origen hipofisario, se realizó cateterismo de los senos petrosos, sin objetivar gradiente entre ellos ni periférico, por lo que se concluyó origen ectópico de la producción de ACTH.
Se practicó una tomografía computarizada toracoabdominal, y se observó una masa en apariencia dependiente del páncreas, homogénea y con pequeñas calcificaciones, de aproximadamente 10 × 7 cm; el resto de la cavidad abdominal era normal. Se realizó un octreoscan con 11In-pentreotida, y se objetivó una gran masa con intensa captación en la cola pancreática.
Con el diagnóstico de tumor neuroendocrino de cola pancreática, se completó el estudio hormonal: gastrina, glucagón, polipéptido pancreático, insulina y polipéptido intestinal vasoactivo, con valores en rango normal.
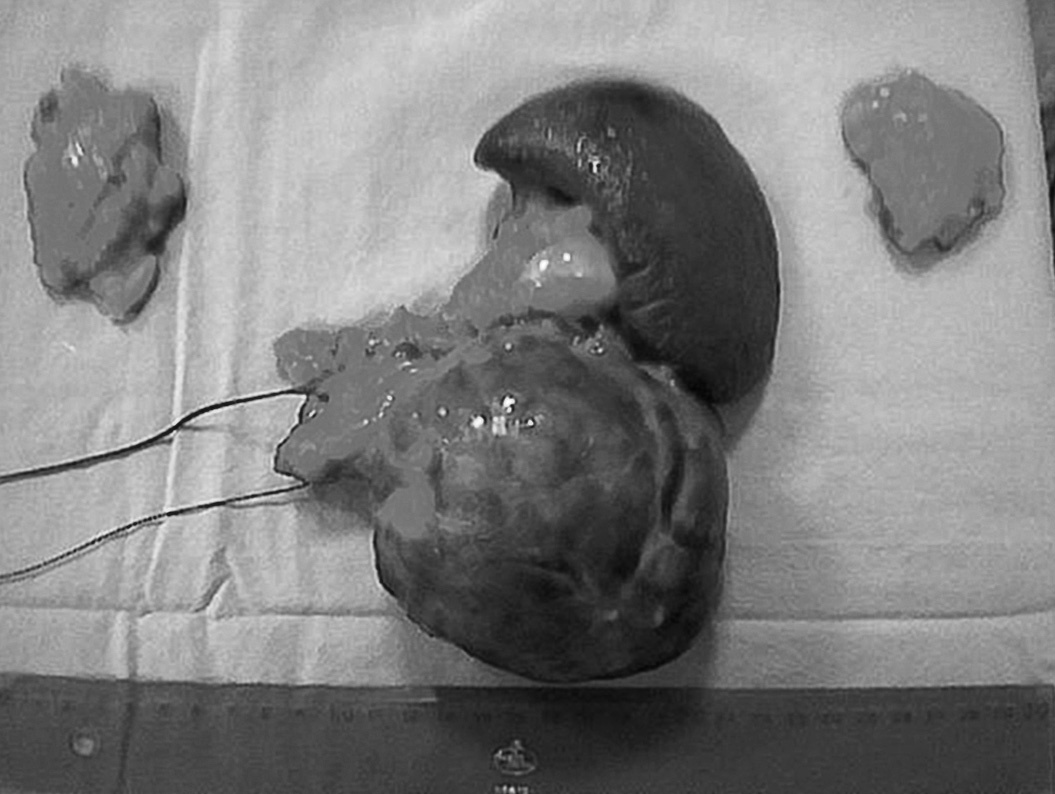
Se decidió intervención quirúrgica y se realizó pancreatectomía distal, esplenectomía y adrenalectomía bilateral (fig. 1). La evolución del paciente fue favorable.
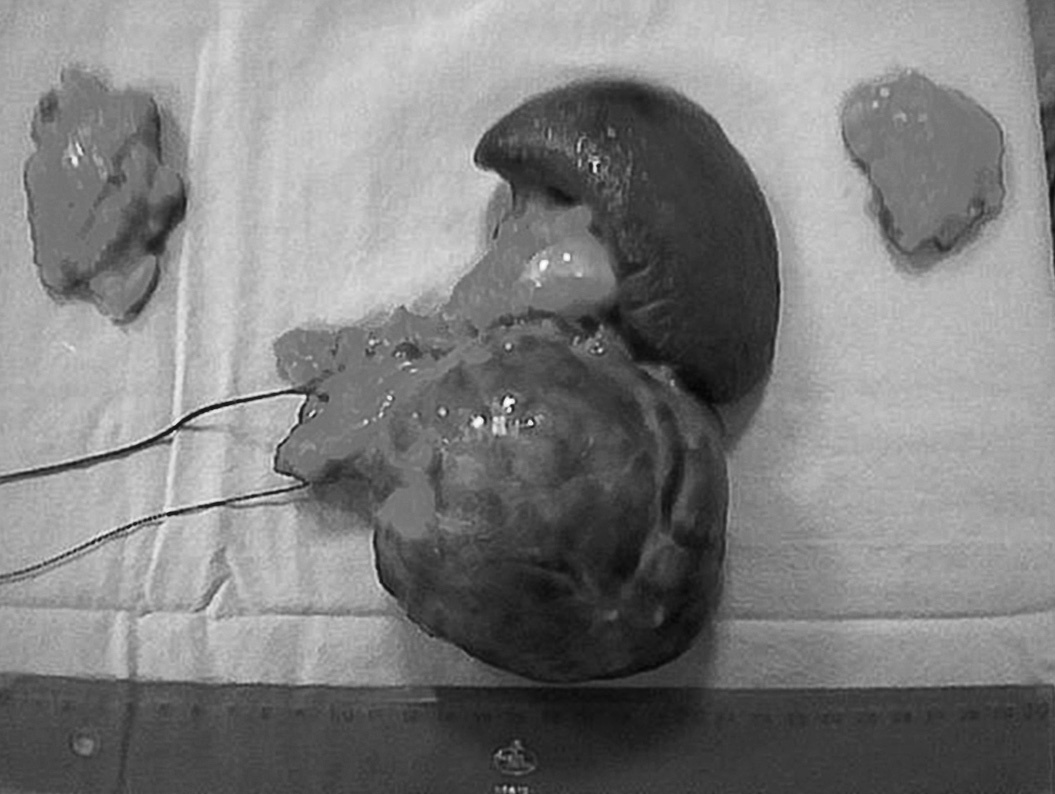
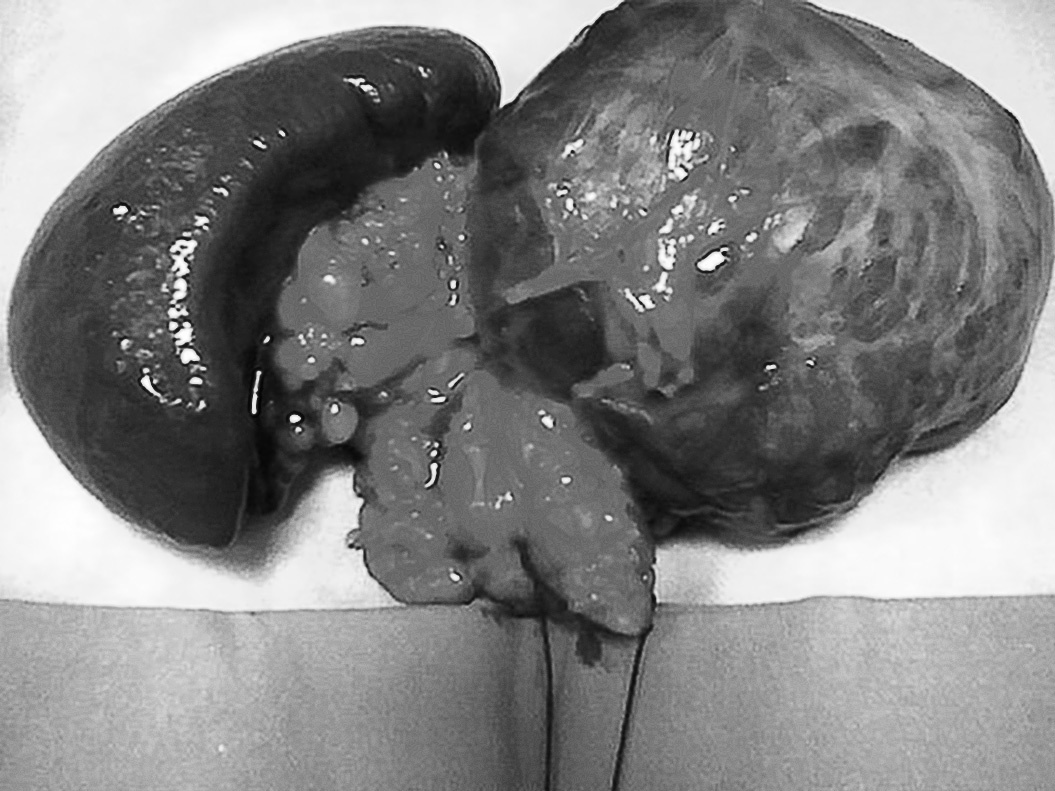
Fig 1. Pieza quirúrgica donde se objetiva el tamaño tumoral, bazo, cola pancreática y ambas suprarrenales.
El resultado del estudio anatomopatológico de la pieza dio como resultado un tumor bien diferenciado de páncreas, de 10 cm de diámetro y bordes quirúrgicos libres de infiltración. Producción de ACTH y glucagón demostrada por inmunohistoquímica.
Los tumores neuroendocrinos del páncreas se originan en las células pluripotenciales localizadas en los conductillos pancreáticos a partir de las cuales se originan los islotes pancreáticos1.
Este tipo de tumores pancreáticos son una rara entidad con un amplio espectro de presentación clínica2.
Aproximadamente, el 50% son no funcionantes; dentro de los funcionantes el más frecuente es el insulinota, seguido del gastrinoma y, con menor frecuencia, aparecen los vipomas, glucagonomas y acthomas1,2,4.
Los tumores pancreáticos productores de ACTH causan menos del 1% de los síndromes de Cushing, y los más frecuentes son los microcíticos de pulmón o los carcinomas bronquiales3,4.
Suelen tener un comportamiento agresivo, con metástasis hepáticas y un gran tamaño con invasión vascular en su presentación clínica3,5.
Pueden asociar producción de otro tipo de hormona, como gastrina; y entonces adquieren un peor pronóstico3,5. En nuestro caso, se tratra de un tumor bien diferenciado con producción asociada de glucagón.
Clínicamente, destaca la ganancia de peso repentina, obesidad central, pérdida de masa muscular, intolerancia a la glucosa e hipertensión1,3.
El diagnóstico del síndrome de Cushing se realiza tras objetivar valores elevados de ACTH plasmático y de cortisol urinario, así como de sus metabólitos (17-hidroxicorticosteroides urinarios). El diagnóstico de localización ectópica se realiza tras objetivar fallo en la supresión del cortisolurinario o de sus metabólitos con altas dosis de dexometasona, ausencia de respuesta adrenal hipofisaria tras la estimulación y aumento de ACTH plasmática sin gradiente en los senos venosos petrosos. Para su localización, la tomografía computarizada y la resonancia magnética son las pruebas que realizar.
Su tratamiento es la cirugía; dado que en el momento del diagnóstico puede presentar metástasis hepáticas o en otras localizaciones más raras, ováricas bilaterales por ejemplo5, la cirugía debe intentar la resección total.
La adrenalectomía bilateral puede ser importante para minimizar las consecuencias del hipercortisolismo intenso y pertinaz en pacientes con tumores neuroendocrinos productores de ACTH que pueden aumentar las complicaciones postoperatorias4.

